基于合成基因和酿酒酵母同源重组机制的分子克隆方法与流程
本申请属于分子克隆技术领域,尤其涉及一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法。
背景技术:
随着技术的发展,人们对于dna的操纵能力逐步提升,对不同大小的质粒进行高效分子克隆的需求也更加多元化。随着测序和dna合成技术的发展,研究者们对于dna的操纵能力大大增强。在基因组合成领域,研究者目前已经成功化学合成了病毒和原核生物的基因组,以及真核生物酿酒酵母的部分染色体。而2016年基因组编写计划(gp-write)则发起针对超大、复杂基因组的拆解-重装-解析的研究,去探索传统方法难以研究的科学问题。在代谢工程领域,利用微生物实现具有重要生理活性和医疗保健作用的天然化合物的全合成已成为世界研究热点,需要进行复杂代谢途径的构建和组装。在这些领域中,研究者们经常需要操纵大量长于10kbp的序列,其中10-150kbp的穿梭质粒(酵母cen-大肠杆菌)使用需求日渐增多。但由于序列长度的原因,这一类质粒的定点修改存在一定的难度,难以实现对其上多区段进行同步深度改造,更遑论多个改造版本的同时构建。
分子克隆技术作为现代生物学的核心技术,被广泛应用于合成生物学、蛋白纯化、生物制药等各个领域,目前有以下方法运用于基因片段的改造,包括:(1)传统的限制性内切酶克隆方法,包括两种不同的方法,一种是利用限制性内切酶特异性地识别目的dna和载体上的酶切位点并进行切割,再通过连接酶,将二者连在一起得到目的质粒,也称为重组载体,但该方法常常出现在需要操作的位置没有可用的酶切位点的情况;另外一种是人为引入新的酶切位点,采用该方法连接起来的两个片段之间会各增加一个酶切位点的6-8bp的碱基片段,这种疤痕序列可能会对目标dna片段产生影响,无法实现无缝拼接。(2)goldengate方法被建立,利用iis型限制性内切酶的切割位点与识别序列分离的特征,通过合理的设计,可使多片段拼接的目的序列中不含有相应的酶切位点,实现多片段的无缝拼接。gibson组装方法被建立,该方法同样适用于多个线性dna片段的无缝拼接,通过为各顺序相邻的dna片段添加20-80bp左右的短重叠序列,同时运用外切酶、聚合酶和dna连接酶实现多dna片段的一步组装。但在片段数目增多和片段长度增加时,以上两种方式的正确拼接效率会大幅度降低。若片段内部存在多个所使用的iis型酶切位点,goldengate方法则更不适用。此外,若使用以上两种方式对10-150kbp的穿梭质粒进行多处序列改造时,需要通过引物扩增出修改后的片段,并引入酶切位点或者同源臂。序列扩增易受序列长度、gc含量以及序列重复度的影响,且对于较长的dna片段,如大于10kbp,容易在序列内部产生非预期的突变。(3)crispr/cas9系统与gibson组装的联合方法被建立,利用crispr/cas9系统在体外对22kbp的质粒进行切割,然后通过pcr扩增插入片段,添加同源序列,通过gibson组装实现了在22kbp质粒载体上任意位点插入783bpdna片段。(4)基于crispr/cas9和酿酒酵母细胞内源的同源重组的分子克隆方法被建立,该方法通过设计特异的靶向grna,利用crispr/cas9体系对初始载体在体外实现一处或多处的切割,回收后与缺口处带有同源序列的被替换dna序列一同转化酵母实现质粒的构建,通过一次转化筛选操作,同时完成将一个或多个目的dna片段的插入或删除。以上方法可进行分子克隆,但存在以下几个主要限制因素:一,需设计和构建特异性靶向待修改位点的grna表达载体,修改位点越多,需要的grna载体数目也越多;二,需购买或自备相应试剂进行sgrna的体外转录和cas9的体外切割;三,克隆成功率受限于grna的特异性以及体外切割体系的效率;四,若需对质粒进行多处修改时,每个版本均需定向制备多个带同源臂序列片段,相对繁琐,难以同时生成多版本的组合突变。目前,针对10-150kbp的穿梭质粒(酵母-大肠杆菌),本领域内仍缺少通用型、可高效实现对中型质粒上多个不连续dna区段同步无缝改造、多种深度改造版本同时构建的分子克隆策略。
技术实现要素:
本申请的目的在于提供一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法,旨在解决现有技术中对于较大穿梭质粒缺乏通用且高效的对单个或多个不连续dna区段同步无缝改造的分子克隆策略的问题。
为实现上述申请目的,本申请采用的技术方案如下:
第一方面,本申请提供一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法,所述分子克隆方法包括如下步骤:
提供待修改载体,将所述待修改载体进行序列比对,确定所述待修改载体的修改区域及替换所述修改区域的标记基因;
设计所述标记基因的第一同源臂引物序列并扩增得到所述标记基因,将所述标记基因片段与所述待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第一质粒前体的酿酒酵母细胞,其中,所述第一质粒前体包括所述标记基因;
合成带有同源臂的供体序列并进行线性化处理得到线性化的供体序列,将所述线性化的供体序列转化至所述含有第一质粒前体的酿酒酵母细胞,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
第二方面,本申请提供一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法,所述分子克隆方法包括如下步骤:
提供待修改载体,将所述待修改载体进行序列比对,确定所述待修改载体的修改区域及替换所述修改区域的标记基因;
设计所述标记基因的第二同源臂引物序列并扩增得到所述标记基因片段,将所述标记基因片段与所述待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第二质粒前体的酿酒酵母细胞,提取所述第二质粒前体并进行富集处理,其中,所述第二质粒前体包括所述标记基因和第二限制性内切酶识别位点;
合成带有同源臂的供体序列,将所述供体序列和所述第二质粒前体分别进行线性化处理得到线性化供体序列和线性化第二质粒前体;
将所述线性化供体序列和所述线性化第二质粒前体共转化至野生型酿酒酵母细胞中,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
本申请第一方面提供的分子克隆方法利用合成基因和酿酒酵母同源重组机制,通过对待修改载体的修改区域进行定位,并设计一对带有同源臂的引物用于标记基因的扩增,再利用酿酒酵母内源的同源重组机制,将标记基因片段对任意待修改的区域进行高效定向取代,获得含有质粒前体的酿酒酵母细胞,实现dna修改的定位,进一步利用基因合成技术生成带同源臂的供体序列,直接将合成片段转化至含质粒前体的酿酒酵母细胞中,利用线性化的合成片段和酵母的同源重组机制实现相应区域的无缝修复,得到含有供体序列质粒的目的酿酒酵母细胞,该分子克隆方法可将多处修改位点合并为一个修改区域,实现在待修改载体的任意位置进行标记基因的插入替代,快速高效获得质粒前体;并结合基因合成技术提供供体序列,利用两端带有同源臂的合成序列与酵母同源重组实现供体序列对标记基因的无缝替代,实现原载体的任意类型(删除、突变或者插入)的修改,不需额外构建其他靶向载体;简单高效可以实现穿梭质粒上多个不连续dna区段的同步改造和多种版本的批量构建。
本申请第二方面提供的分子克隆方法利用合成基因和酿酒酵母同源重组机制,通过对待修改载体的修改区域进行定位,提供含有同源臂的引物序列扩增得到标记基因片段,将标记基因片段对任意待修改的区域进行高效定向取代,筛选得到包括标记基因和第二限制性内切酶识别位点的质粒前体,利用酿酒酵母内源的同源重组机制在质粒前体中引入限制性内切酶识别位点,在后续试验中,只需采用限制性内切酶进行体外酶切和酵母转化实现质粒构建,操作十分简单,不需要sgrna的体外转录和cas9的体外切割的试剂盒;同时对质粒前体进行富集,得到大量质粒前体,有利于后续进行酶切处理;进一步利用基因合成技术提供带同源臂的供体序列,利用酶切的方法分别将供体序列和富集得到的质粒前体进行体外线性化处理后,共转入酿酒酵母细胞中,利用质粒前体与供体序列的体外线性化和酵母同源重组,实现待修改载体序列的任意类型的无缝修改,批量、高效地无缝构建多版本含有供体序列质粒,其中将构建得到的质粒前体进行富集后再进行线性化处理,可降低目的质粒构建时的假阳性率,提高含供体序列的目的质粒的构建成功率。该分子克隆方法将多处修改位点并为修改区域,片段准备时只需对包括替换了修改区域的标记基因片段和限制性内切酶识别位点的质粒前体和合成的供体片段利用限制性内切酶进行体外酶切,再进行共转化至酿酒细胞中,操作十分简单,构建效率较高,构建成功率较高,使得本发明适用于多版本、高通量的无缝克隆操作。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
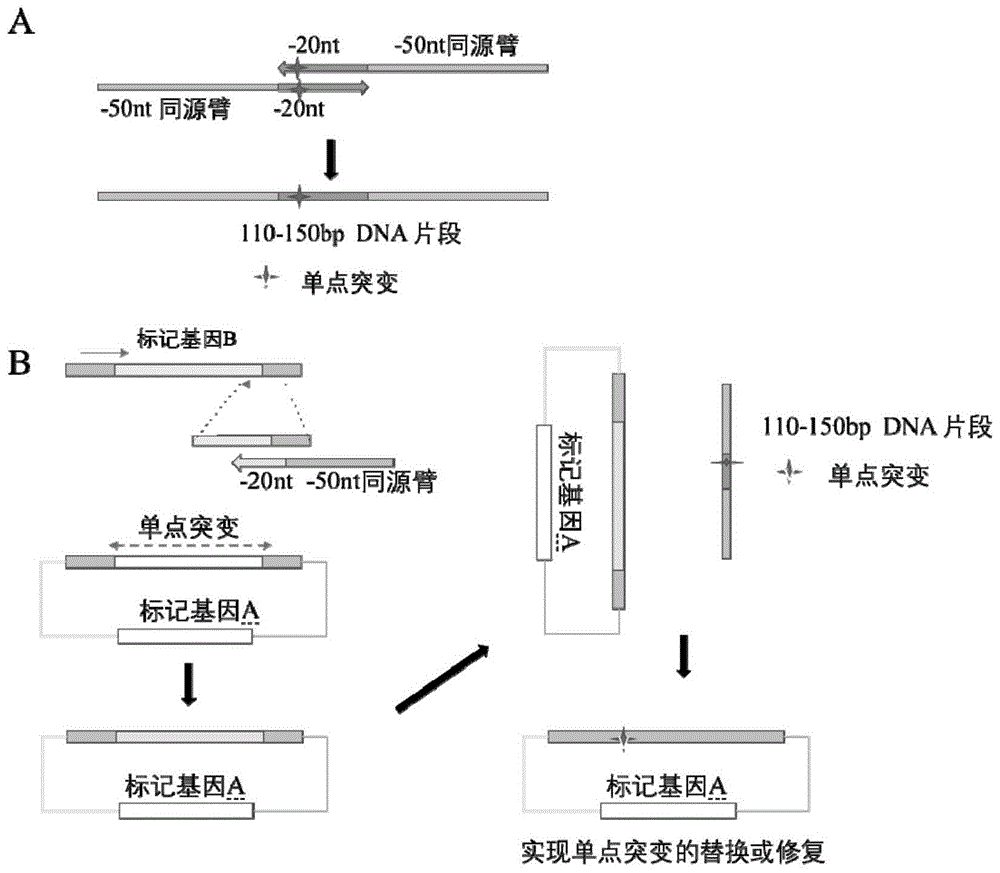
图1是本申请实施例1提供分子克隆的方法的原理及主要流程图。
图2是本申请实施例1提供的pjs379的原载体图谱。
图3是本申请实施例1提供的第一质粒前体的阳性克隆子的菌落pcr结果。
图4是本申请实施例1提供的供体序列的pcr结果。
图5是本申请实施例1提供的转化子的菌落pcr验证结果。
图6是本申请实施例1提供的阳性转化子的高保真dna聚合酶pcr结果。
图7是本申请实施例1提供的阳性转化子高保真dna聚合酶pcr扩增产物测序结果。
图8是本申请实施例2提供分子克隆的方法的原理及主要流程图。
图9是本申请实施例2提供的pjs380的原载体图谱。
图10是本申请实施例2提供的可能含有第二质粒前体的酵母转化子的菌落pcr验证结果。
图11是本申请实施例2提供的待修改区域s4、标记基因ura3片段、供体序列sg016、供体序列sg017、修改后的s4-v1(含有sg016的供体序列质粒)、修改后的s4-v2(含有sg017的供体序列质粒)的序列比对结果。
图12是本申请实施例2提供的第二质粒前体、供体序列sg016、供体序列sg017的线性化处理结果。
图13是本申请实施例2提供的修改后的s4-v1(可能含有sg016的供体序列质粒)的酵母转化子的菌落pcr验证结果。
图14是本申请实施例2提供的修改后的s4-v2(可能含有sg017的供体序列质粒)的酵母转化子的菌落pcr验证结果。
具体实施方式
为了使本申请要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本申请进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本申请,并不用于限定本申请。
应理解,在本申请的各种实施例中,上述各过程的序号的大小并不意味着执行顺序的先后,部分或全部步骤可以并行执行或先后执行,各过程的执行顺序应以其功能和内在逻辑确定,而不应对本申请实施例的实施过程构成任何限定。
在本申请实施例中使用的术语是仅仅出于描述特定实施例的目的,而非旨在限制本申请。在本申请实施例和所附权利要求书中所使用的单数形式的“一种”、“所述”和“该”也旨在包括多数形式,除非上下文清楚地表示其他含义。
本申请实施例第一方面提供一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法,分子克隆方法包括如下步骤:
s01.提供待修改载体,将待修改载体进行序列比对,确定待修改载体的修改区域及替换修改区域的标记基因;
s02.设计标记基因的第一同源臂引物序列并扩增得到标记基因片段,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第一质粒前体的酿酒酵母细胞,其中,第一质粒前体包括标记基因;
s03.合成带有同源臂的供体序列并进行线性化处理得到线性化的供体序列,将线性化的供体序列转化至含有第一质粒前体的酿酒酵母细胞,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
本发明提供一种结合基因合成和酿酒酵母内源同源重组系统的分子克隆方法,不受限于序列改变的类型(删除、突变或者插入)和复杂度,仅涉及简单的体外分子克隆操作,利用两端带有同源臂的合成序列与酵母同源重组实现原载体的无缝修改,可实现4-150kbp穿梭质粒上多个不连续dna区段的同步改造和多种版本的批量构建。
具体的,在上述步骤s01中,提供待修改载体,将待修改载体进行序列比对,确定待修改载体的修改区域及替换修改区域的标记基因。
其中,待修改载体指可实施目的片段的删除、突变或插入的载体。优选的,待修改载体选自大肠杆菌-酿酒酵母的穿梭载体,使该载体能够在该分子克隆方法中进行穿梭试验,能够保证在酿酒酵母细胞中复制进而富集载体,同时也能够在大肠杆菌中大量复制进而富集载体。
进一步优选的,待修改载体还包括在其他细胞中进行穿梭使用。在一些实施例中,待修改载体包括但不限于大肠杆菌-酿酒酵母的穿梭载体、大肠杆菌-酿酒酵母-链霉菌的穿梭载体、大肠杆菌-酿酒酵母-枯草芽孢杆菌的穿梭载体、大肠杆菌-酿酒酵母-谷氨酸棒状杆菌的穿梭载体、大肠杆菌-酿酒酵母-丝状真菌的穿梭载体、大肠杆菌-酿酒酵母-哺乳动物细胞的穿梭载体、大肠杆菌-酿酒酵母-植物细胞的穿梭载体。
在一些实施例中,待修改载体选自环状质粒载体。
在一些实施例中,待修改载体包括但不限于空载体或携带目的基因的重组载体。所提供的分子克隆方法适用于各类型的载体改造,可用于空载体中某些标记基因的片段修改,也可用于携带目的基因的重组载体的标记基因或目的基因的修改,简单高效。
优选的,待修改载体的碱基数为4~150kbp,本申请提供的分子克隆方法适用于碱基数为4~150kbp的穿梭质粒的改造。
进一步的,将待修改载体进行序列比对,确定待修改载体的修改区域及替换修改区域的标记基因。将提供的待修改的载体与现有的序列进行序列比对,进而确定待修改载体的修改区域;并提供替换修改区域的标记基因。
在一些实施例中,修改区域包括但不限于一个或多个dna片段。在一些实施例中,修改区域包括但不限于多个不连续dna片段。
在一些实施例中,标记基因包括但不限于一个或多个标记基因。
优选的,替换修改区域的标记基因中,可将待修改载体上一个或多个(包括但不限于1个、2个、3个、4个、5个、6个、7个、8个、9个、10个或以上)的dna片段替换为一个标记基因。在一些实施例中,多个不连续的dna片段合并成一个dna片段区域替换为一个标记基因。在本发明具体实施例中,针对某一修改区域而言,对替换的标记基因的片段数量没有特别的限制。优选的,标记基因包括但不限于编码抗生素抗性基因、营养缺陷相关基因、荧光蛋白或融合荧光蛋白。在一些实施例中,标记基因包括但不限于ura3、trp1、his3、lys2、leu2或ade2的营养缺陷型基因片段。在一些实施例中,标记基因包括但不限于kanmx4或hygmx4、natmx4的编码抗生素抗性基因。
具体的,在上述步骤s02中,设计标记基因的第一同源臂引物序列,并扩增得到标记基因片段,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第一质粒前体的酿酒酵母细胞,其中,第一质粒前体包括标记基因。
其中,本发明采用酿酒酵母细胞内源的同源重组系统实现标记基因片段对修改载体的修改区域的替换,并利用标记基因筛选重组子。同源重组是指发生在含有同源序列的dna分子之间或分子之内的重新组合。优选的,设计标记基因的第一同源臂引物序列的步骤中,第一同源臂引物序列从5’-3’端依次包括修改区域两侧的同源臂序列以及目的dna的扩增引物序列,设计带有修改区域两侧的同源臂序列,同源臂序列用于识别并发生重组的区域使标记基因通过酵母转化,利用同源臂序列替换待修改载体中的修改区域。进一步优选的,同源臂序列的长度为45~50nt。
在一些实施例中,利用第一同源臂引物序列作为标记基因的扩增引物,以含有标记基因的其他载体作为模板进行pcr扩增,得到标记基因片段。
在一些实施例中,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞的步骤中,包括但不限于选择醋酸锂转化法、电转化法或原生质体转化法将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞。
在一些实施例中,筛选得到含有第一质粒前体的酿酒酵母细胞的步骤中,选择第一质粒前体带有的与待修改载体不同的抗生素抗性基因或营养缺陷筛选标记基因进行筛选,从而转化子能够在添加有抗生素或不含某种必须营养成分的选择性培养基上生长,由于待修改载体上不含这些选择标记,因此,不含任何载体的酵母转化子或不含标记基因的待修改载体的酵母转化子均不能够在选择性培养基上生长,通过该方法筛选得到含有第一质粒前体的酿酒酵母细胞。
进一步的,第一质粒前体包括标记基因;标记基因利用酿酒酵母内源的同源重组机制,将标记基因片段对任意待修改的区域进行高效定向取代,获得含有质粒前体的酿酒酵母细胞,实现dna修改的定位。
具体的,在上述步骤s03中,合成带有同源臂的供体序列并进行线性化处理得到线性化的供体序列,将线性化的供体序列转化至含有第一质粒前体的酿酒酵母细胞,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
优选的,供体序列的两端包括第一限制性内切酶识别位点,所添加第一限制性内切酶识别位点为了使供体序列利用线性化处理,方便转化至含有第一质粒前体的酿酒酵母细胞。进一步优选的,第一限制性内切酶识别位点为供体序列内不存在的限制性内切酶识别位点。
进一步,将线性化的供体序列转化至含有第一质粒前体的酿酒酵母细胞的步骤中,包括但不限于选择醋酸锂转化法、电转化法或原生质粒转化法将线性化的供体序列转化至含有第一质粒前体的酿酒酵母细胞。
进一步,筛选得到含有供体序列质粒的目的酿酒酵母细胞的步骤中,包括但不限于采用多重pcr、抗生素抗性培养基或营养缺陷培养基进行筛选。
优选的,分子克隆方法还包括:从含有供体序列质粒的目的酿酒酵母细胞中提取含有供体序列质粒,将含有供体序列质粒转化至大肠杆菌中进行富集处理。由于提供的待修改质粒为大肠杆菌-酿酒酵母的穿梭载体,因此将含有供体序列质粒转化至大肠杆菌中进行富集处理并进行酶切鉴定、测序分析,得到纯度高、浓度高的供体序列质粒,有利于进行后续生物学相关研究。
利用基因合成技术生成带同源臂的供体序列,直接将合成片段转化至含质粒前体的酿酒酵母细胞中,利用线性化的合成片段和酵母的同源重组机制实现相应区域的无缝修复,得到含有供体序列质粒的目的酿酒酵母细胞,该方法快速简便,可实现原载体的无缝修改,不需额外构建其他靶向载体。
本申请实施例第二方面提供一种基于合成基因和酿酒酵母同源重组机制的分子克隆方法,分子克隆方法包括如下步骤:
g01.提供待修改载体,将待修改载体进行序列比对,确定待修改载体的修改区域及替换修改区域的标记基因;
g02.设计标记基因的第二同源臂引物序列并扩增得到标记基因片段,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第二质粒前体的酿酒酵母细胞,提取第二质粒前体并进行富集处理,其中,第二质粒前体包括标记基因和第二限制性内切酶识别位点;
g03.合成带有同源臂的供体序列,将供体序列和第二质粒前体分别进行线性化处理得到线性化供体序列和线性化第二质粒前体;
g04.将线性化供体序列和线性化第二质粒前体共转化至野生型酿酒酵母细胞中,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
本申请第二方面提供的分子克隆方法利用合成基因和酿酒酵母同源重组机制,通过在设计的引物上增加限制性内切酶的识别位点;同时对质粒前体进行富集,得到大量含有标记基因的质粒前体,将含有标记基因的质粒前体与合成的供体序列进行体外线性化后共转化,利用线性的质粒前体与供体序列和酵母同源重组,实现待修改载体序列的任意类型的无缝修改,批量、高效地无缝构建多版本含有供体序列质粒,提高供体序列质粒的构建成功率,可快速、高效进行质粒载体的克隆。
具体的,在上述步骤g01中,提供待修改载体,将待修改载体进行序列比对,确定待修改载体的修改区域及替换修改区域的标记基因。此处,g01所论述的内容与上述s01所论述的内容一致,为了节约篇幅,此处不再进行赘述。
优选的,待修改载体选自大肠杆菌-酿酒酵母的穿梭载体,使该载体能够在该分子克隆方法中进行穿梭试验,能够保证在酿酒酵母细胞中稳定存在,同时也能够在大肠杆菌中大量复制进而富集载体。
优选的,待修改载体的碱基数为4~150kbp,本申请第二方面提供的分子克隆方法适用于碱基数为4~150kbp的穿梭质粒的改造。进一步优选的,待修改载体的碱基数为20~30kbp,针对碱基数为20~30kbp的待修改载体,采用本申请第二方面提供的分子克隆方法进行改造,操作十分简单,构建效率高,构建成功率高。
具体的,在上述步骤g02中,设计标记基因的第二同源臂引物序列并扩增得到标记基因片段,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞,筛选得到含有第二质粒前体的酿酒酵母细胞,提取第二质粒前体并进行富集处理,其中,第二质粒前体包括标记基因和第二限制性内切酶识别位点。
优选的,设计标记基因的第二同源臂引物序列的步骤中,第二同源臂引物序列从5’-3’端依次包括修改区域两侧的同源臂序列、第二限制性内切酶识别位点以及标记基因的扩增引物序列,其中,设计带有修改区域的两侧同源臂序列,同源臂序列用于识别并发生重组的区域使标记基因通过酵母转化,利用同源臂序列替换待修改载体中的修改区域;进一步,还包括第二限制性内切酶识别位点,在第二质粒前体中引入第二限制性内切酶识别位点,有利于在后续试验中,对富集后的、纯度高的第二质粒前体后进行体外酶切,利用质粒前体与供体序列的体外线性化和酵母同源重组,实现多版本目的质粒的批量、高效的无缝构建,提高载体的构建成功率,同时避免后续步骤中出现较多的假阳性,影响正确率。
进一步优选的,第二限制性内切酶识别位点为待修改载体上不存在的酶切位点,确保进行特异性地线性化处理,有利于供体序列与质粒前体成功组装。
进一步优选的,同源臂序列的长度为45~50nt。
进一步,将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞的步骤中,包括但不限于选择醋酸锂转化法、电转化法、原生质体转化法将标记基因片段与待修改载体共转化至酿酒酵母细胞。
进一步,筛选得到含有第二质粒前体的酿酒酵母细胞的步骤中,选择第二质粒前体带有的与待修改载体不同的抗生素抗性基因或营养缺陷筛选标记基因进行筛选,从而转化子能够在添加有抗生素或不含某种必须营养成分的选择性培养基上生长,由于待修改载体上不含这些选择标记,因此,不含任何载体的酵母转化子或不含标记基因的待修改载体的酵母转化子均不能够在选择性培养基上生长,通过该方法筛选得到含有第二质粒前体的酿酒酵母细胞。在一些实施例中,进一步采用菌落pcr的验证方法筛选含有第二质粒前体的酿酒酵母细胞。
进一步,第二质粒前体包括标记基因和第二限制性内切酶识别位点,通过在第二同源臂引物序列中设计带有第二限制性内切酶识别位点,实现在第二质粒前体中设置标记基因和第二限制性内切酶识别位点,经过对第二质粒前体进行提取、富集和体外酶切后,有利于与供体序列进行高效重组,确保后续的转化组装正确率较高,且不会出现较多的假阳性。
优选的,提取第二质粒前体并进行富集处理的步骤中,包括:从含有第二质粒前体的酿酒酵母细胞中提取第二质粒前体,将第二质粒前体转化至大肠杆菌中进行单克隆纯化和富集处理。由于提供的待修改质粒为包括大肠杆菌-酿酒酵母的穿梭载体,因此将第二质粒前体转化至大肠杆菌中进行富集处理,得到纯度高、浓度高的第二质粒前体,有利于进行后续的酶切线性化与酵母转化试验,提高正确率。
具体的,在上述步骤g03中,合成带有同源臂的供体序列,将供体序列和第二质粒前体分别进行线性化处理得到线性化供体序列和线性化第二质粒前体。优选的,供体序列的两端包括第一限制性内切酶识别位点,所添加第一限制性内切酶识别位点为了将供体序列线性化。进一步优选的,第一限制性内切酶识别位点为供体序列内部不存在的限制性内切酶识别位点。进一步,采用限制性内切酶对富集得到的第二质粒前体进行线性化处理得到线性化第二质粒前体,采用限制性内切酶对供体序列进行线性化处理得到线性化供体序列。
具体的,在上述步骤g04中,将线性化供体序列和线性化第二质粒前体共转化至野生型酿酒酵母细胞中,筛选得到含有供体序列质粒的目的酿酒酵母细胞。利用质粒前体与供体序列的体外线性化和酵母同源重组,实现待修改载体序列的任意类型的无缝修改,批量、高效地无缝构建多版本含有供体序列质粒,其中将构建得到的质粒前体进行富集后再进行线性化处理,提高供体序列质粒的构建成功率,可快速、高效进行质粒载体的克隆。
进一步,将线性化供体序列和线性化第二质粒前体共转化至野生型酿酒酵母细胞中,包括但不限于选择醋酸锂转化法、电转化法或原生质体转化法将线性化供体序列和线性化第二质粒前体共转化至野生型酿酒酵母细胞中。将序列拼接转变成的酵母细胞内两个线性序列的组装,实现待修改载体序列的任意类型的无缝修改,批量、高效地无缝构建多版本含有供体序列质粒,提高供体序列质粒的构建成功率,可快速、高效进行质粒载体的克隆。
进一步,筛选得到含有供体序列质粒的目的酿酒酵母细胞的步骤中,包括但不限于采用多重pcr、抗生素抗性培养基或营养缺陷培养基进行筛选。在一些实施例中,进一步采用菌落pcr的验证方法筛选含有第二质粒前体的酿酒酵母细胞。
优选的,分子克隆方法还包括:从含有供体序列质粒的目的酿酒酵母细胞中提取含有供体序列质粒,将含有供体序列质粒转化至大肠杆菌中进行富集处理。由于提供的待修改质粒为包括大肠杆菌-酿酒酵母的穿梭载体,因此将含有供体序列质粒转化至大肠杆菌中进行富集处理并进行酶切鉴定、测序分析,得到纯度高、浓度高的供体序列质粒,有利于进行后续生物学相关研究。
该分子克隆方法将多处修改位点并为修改区域,片段准备时只需对包括替换了修改区域的标记基因片段和限制性内切酶识别位点的质粒前体和合成的供体片段利用限制性内切酶进行体外酶切,再进行共转化至酿酒细胞中,操作十分简单,构建效率较高,构建成功率较高,使得本发明适用于多版本、高通量的无缝克隆操作。
下面结合具体实施例进行说明。
实施例1
一种基于合成基因和酿酒酵母同源重组机制的pjs379的分子克隆方法
一种基于合成基因和酿酒酵母同源重组机制的pjs379的分子克隆方法(本申请第一方面提供的分子克隆方法利用合成基因和酿酒酵母同源重组机制)的原理和主要过程如附图1所示,具体步骤如下:
(1)提供待修改载体pjs379,其中,pjs379,为大肠杆菌-酿酒酵母的穿梭质粒,能够在大肠杆菌细胞和酿酒酵母细胞中实现自主复制和在含有相应的筛选条件下生长,携带有氨苄青霉素抗性基因、酵母his3标记基因(标记基因a),还承载了多个哺乳动物基因表达单元,原始质粒大小为29.2kbp,如附图2所示。
将待修改载体pjs379进行比对分析,确定待修改载体的修改区域为s2基因序列上包含待修改碱基的50bp左右的区域;将经过分子实验后实现单碱基替换的质粒命名为s2-mut。
进一步,选定替换修改区域的标记基因为ura3。
(2)①设计标记基因ura3的第一同源臂引物序列:其中,第一同源臂引物序列从5’-3’端依次包括修改区域的两侧同源臂序列以及标记基因的扩增引物序列;具体的,标记基因ura3的第一同源臂引物序列为jso1415与jso1416,其中,jso1415的序列如seq.idno.1(其中,加粗的碱基序列为同源臂序列):
jso1416的序列如seq.idno.2(其中,加粗的碱基序列为同源臂序列):
②扩增标记基因ura3序列:以酵母整合型载体prs406为模板,以jso1415与jso1416为扩增引物,扩增得到带同源臂的标记基因ura3-1片段,其中,带同源臂的标记基因ura3-1片段的长度为1276bp。
dnaura3-1片段的序列如seq.idno.3,seq.idno.3的具体碱基序列如序列表中seq.idno.3所示。
③将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞:采用醋酸锂转化法将标记基因ura3片段与待修改载体pjs379共转化酿酒酵母细胞by4742中。其中,转化过程如下:将酿酒酵母by4742单菌落接种到ypd培养基中于30℃过夜培养后,转接至新的ypd液体培养基中;将起始菌体浓度调整为od600=0.1,于30℃、220rpm条件下培养4-5小时至菌体浓度在od600为0.4-0.5;收集5ml菌体于4℃、3000rpm条件下离心5min并收集第一细胞沉淀,用5mlddh2o重悬细胞沉淀后于4℃、3000rpm条件下离心5min并收集第二细胞沉淀,利用1ml0.1mol/llioac/te溶液重悬第二细胞沉淀后转移到1.5mlep管中,于4℃、3000rpm条件下离心5min收集第三细胞沉淀,利用100μl0.1mol/llioac/te溶液重悬第三细胞沉淀,得到酵母感受态细胞;将待转化的标记基因ura3片段与待修改载体pjs380加入到酵母感受态细胞得到第一酵母感受态细胞中;在制备感受态细胞过程中,同步将鲑鱼精dna(ssdna)于100℃条件下处理5分钟并迅速置于冰上冷却备用;提供312μl质量百分浓度为50%peg3350转化液、41μl1mol/llioac溶液、25μl10mg/ml鲑鱼精dna(ssdna)加入到含有酵母感受态和转化dna的ep管中,振荡混匀后,于30℃孵育45分钟得到第一混合细胞;在第一混合细胞加入50μldmso,于42℃水浴热击15分钟后,于3000rpm离心1min收集第一混合细胞沉淀;采用500μl5mmol/l的cacl2溶液清洗第一混合细胞沉淀,于3000rpm离心1min收集第二混合细胞沉淀,加入200μlddh2o重悬第二混合细胞沉淀得第三混合细胞溶液。
④筛选得到含有第一质粒前体的酿酒酵母细胞:将第三混合细胞溶液涂布到sc-ura的平板上,放置于30℃培养箱中培养48小时,并挑取部分菌落进行菌落pcr验证,筛选得到含有第一质粒前体的酿酒酵母细胞。
其中,菌落pcr验证步骤中,采用验证引物jso1040和验证引物12311进行验证,验证引物jso1040的序列如seq.idno.4,seq.idno.4如下所示:cccaacaggtctcttgatggtc;筛选引物12311的序列如seq.idno.6,seq.idno.6如下所示:ctgtgctccttccttcgttc。
进一步,采用验证引物jso1221和引物jso1419进行筛选,验证引物jso1221的序列如seq.idno.5,seq.idno.5如下所示:cgtggatgatgtggtctctacagg;验证引物jso1419的序列如seq.idno.9,seq.idno.9如下所示:cttgcaatattcctacctggtgtctc。
(3)①采用两个长引物构建带有同源臂的供体序列,将替换的碱基设计在两条长引物中,这两条长引物分别带有修改区域左右两侧的同源臂,50nt左右的同源序列,单点突变点在两条长引物的重叠附近区域。
提供长引物jso1417与jso1418,长引物jso1417的序列如seq.idno.7,seq.idno.7如下所示(其中,加粗和下划线的碱基“t”为目的替换的碱基):
长引物jso1418的序列如seq.idno.8,seq.idno.8如下所示
5’-agtggtaagggtttacacatacttcatcctttttaagattaaaagcatattcgcagttttcgatagctttcagctcgtggtg-3’。
通过pcr程序进行退火配对后补齐,将两条单链引物拼接为一条短链的dna片段,得到供体序列。
②将线性化的供体序列转化至含有第一质粒前体的酿酒酵母细胞:将线性化供体序列转化至含有第一质粒前体的酿酒酵母细胞中,转化过程如上述(2)③。
③筛选得到含有供体序列质粒的目的酿酒酵母细胞:将转化了供体序列和第一质粒前体的酿酒酵母细胞涂布于sc-his平板,于30℃条件下培养24小时得到第一平板菌落;将第一平板菌落影印至sc-his+5-foa(1g/l)平板、sc-ura及平板上,于30℃条件48小时,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定,筛选得到含有供体序列质粒的目的酿酒酵母细胞。
其中,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定的步骤中,采用菌落pcr鉴定引物jso1040和菌落pcr鉴定引物jso1419进行鉴定,菌落pcr鉴定引物jso1040的序列如seq.idno.4,seq.idno.4如下所示:cccaacaggtctcttgatggtc;菌落pcr鉴定引物jso1419的序列如seq.idno.9,seq.idno.9如下所示:cttgcaatattcctacctggtgtctc。
进行菌落pcr鉴定的菌落pcr反应体系如下:
挑取待检测菌落到20μl20mmnaoh中,置于pcr仪中,(98℃3min,4℃2min),5个循环,后静置备用;
再按照下表1的菌落pcr反应体系进行配制混合溶液,按照下表2所示的pcr反应程序进行pcr反应。
表1
表2
④pcr测序:由于点突变,故最终需要经过测序确定是否为阳性菌落,选取初步阳性菌落用高保真酶扩增覆盖点突变区域的dna片段,并送pcr产物测序,方能最终确定阳性克隆子。
结果分析:
实施例1的结果分析如下:
结果分析(一)
根据实施例1步骤(2)④中,将第三混合细胞溶液涂布到sc-ura的平板上,放置于30℃培养箱中培养48小时,并挑取16个菌落进行菌落pcr验证,菌落pcr验证结果如图3所示,可以发现13个菌落均有两个阳性条带,片段大小分别为245bp和347bp,为阳性克隆子,均为含有第一质粒前体的酿酒酵母细胞,进一步进行阳性率统计,如下表3所示,转化子为181个,阳性克隆比为13/16,阳性率为81.25%。
表3
结果分析(二):
根据实施例1步骤(3)①中,供体序列如图4所示,图4(1、2)为采用两个长引物构建带有同源臂的供体序列,含有s2单点突变引物经重叠pcr产物合成片段的pcr结果,片段大小为143bp。
结果分析(三)
根据实施例1步骤(3)③,其中,sc-his+5-foa平板是为了在筛选正确重组质粒的细胞的同时,除去胞内还同时含有第一质粒前体的假阳性酵母菌;sc-ura板主要用于推断假阳性酵母菌的大致比例。实验发现生长在sc-ura平板上的菌落非常多,而能够在sc-his+5-foa平板上的酵母菌落数较少,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定,筛选得到含有供体序列的目的酿酒酵母细胞的步骤中,如图5所示,可以发现,验证的84个转化子中,51个菌落均有阳性条带,片段大小为218bp,为阳性克隆子,进一步进行阳性率统计,如下表4所示,转化子为84个,阳性克隆比为51/84,阳性率为60.7%。
表4
进一步采用高保真dna聚合酶对s2-mut初步阳性菌落扩增含突变区域进行pcr扩增,结果如图6所示,a、b、c、d均为阳性克隆子,片段大小为218bp,进一步进行测序分析。
结果分析(四)
随机从上述四个阳性克隆子的pcr产物中选取两个进行测序分析,测序结果如图7所示,2个pcr产物测序结果与目标一致,实现了正确修复。
通过实施例1证明本发明提供的分子克隆方法将质粒pjs379进行了单点改造,该分子改造的方法操作十分简单,构建效率较高,不需额外构建其他靶向载体。该方法更为适用于没有合适的第二限制性酶切位点及质粒大小太大而不容易提取的情况,通过该方法的得到的转化子可能较少,但最终的构建效率较高,过程简单。
实施例2
一种基于合成基因和酿酒酵母同源重组机制的pjs380的分子克隆方法
一种基于合成基因和酿酒酵母同源重组机制的pjs380的分子克隆方法(本申请第二方面提供的分子克隆方法利用合成基因和酿酒酵母同源重组机制)的原理和主要过程如附图8所示,具体步骤如下:
(1)提供待修改载体pjs380,其中,pjs380,为大肠杆菌-酿酒酵母的穿梭质粒,能够在大肠杆菌细胞和酿酒酵母细胞中实现自主复制和在含有相应的筛选条件下生长,携带有氨苄青霉素抗性基因、酵母his3标记基因(标记基因a),还承载了多个哺乳动物基因表达单元,该质粒长度为27.29kbp,如图9所示。
将待修改载体pjs380进行比对分析,确定待修改载体的修改区域为s4,且修改区域分布在5.7kbp的区域内;
进一步,选定替换修改区域的标记基因b为ura3。
(2)①设计标记基因ura3的第二同源臂引物序列:其中,第二同源臂引物序列从5’-3’端依次包括修改区域两侧的同源臂序列、第二限制性内切酶识别位点以及标记基因的扩增引物序列;通过序列分析,选择srfi识别位点作为第二限制性内切酶;具体的,标记基因ura3的第二同源臂引物序列为jso1620与jso1621,其中,jso1620的序列如seq.idno.10(其中,加粗的碱基序列为同源臂序列,下划线的序列为限制性内切酶srfi的识别序列):
jso1621的序列如seq.idno.11(其中,加粗的碱基序列为同源臂序列,下划线的序列为限制性内切酶srfi的识别序列):
②扩增标记基因ura3序列:以酵母整合型载体prs406为模板,以jso1620与jso1621为扩增引物,扩增得到带同源臂的标记基因ura3-2片段,其中,带同源臂的标记基因ura3-2片段的长度为1288bp。
dnaura3-2片段的序列如seq.idno.12,seq.idno.12的具体碱基序列如序列表中seq.idno.12的序列所示。
③将标记基因片段b与待修改载体共转化至酿酒酵母细胞:采用醋酸锂转化法将标记基因dnaura3片段与待修改载体pjs380共转化酿酒酵母细胞by4742中。其中,转化过程与实施例1(2)③中“将标记基因片段与待修改载体共转化至野生型酿酒酵母细胞:采用醋酸锂转化法将标记基因ura3片段与待修改载体pjs379共转化酿酒酵母细胞by4742中”的转化过程一致,此处不再进行赘述。④筛选得到含有第二质粒前体的酿酒酵母细胞:将第三混合细胞溶液涂布到sc-ura的平板上,放置于30℃培养箱中培养48小时,并挑取部分菌落进行菌落pcr验证,采用下表5提供的pcr反应体系进行配制混合溶液,按照下表6所示的pcr反应程序进行pcr反应,筛选得到含有第二质粒前体的酿酒酵母细胞。
其中,菌落pcr验证步骤中,采用验证引物jso1061和验证引物12311进行验证,验证引物jso1061的序列如seq.idno.13,seq.idno.13如下所示:gacaagttggcagcaacaacac;验证引物12311的序列如seq.idno.6,seq.idno.6如下所示:ctgtgctccttccttcgttc。
筛选得到含有第二质粒前体的酿酒酵母细胞的中,采用筛选引物jso1221和筛选引物jso1521进行验证,验证引物jso1221的序列如seq.idno.5,seq.idno.5如下所示:
cgtggatgatgtggtctctacagg;验证引物jso1521的序列如seq.idno.14,seq.idno.14如下所示:ctgaagttgcaagacaatctgaattg。
⑤提取第二质粒前体并进行富集处理:挑取含有第二质粒前体的酿酒酵母细胞菌落进行培养,并应用酵母质粒提取方法提取第二质粒前体,将第二质粒前体转化至大肠杆菌感受态细胞中,涂布到氨苄平板上;对转化子进行pcr初步验证,挑选鉴定正确的菌落进行培养后利用商业常规质粒提取试剂盒将质粒前体提取出来,酶切鉴定,并将质粒前体上包含标记基因及两侧同源臂的序列进行测序鉴定。通过培养大肠杆菌阳性克隆,大量富集,得到纯度高、浓度大的第二质粒前体,其中,第二质粒前体包括标记基因b的dna序列和第二限制性内切酶识别位点。
具体过程如下:将含有第二质粒前体的酿酒酵母细胞单菌落接种到sc-ura培养基培养约16小时,于3000rpm离心5min后收集第一菌体沉淀;用840μlspebuffer(1msorbitol,0.01msodiumphosphate,0.01mna2-edta(ph7.5))重悬第一菌体沉淀,加入1μl巯基乙醇和1μl20mg/mlzymolyase100t溶液混匀,放置于37℃培养箱中1小时得到第二菌体溶液,每间隔10min上下颠倒混匀一次;将第二菌体溶液于3000rpm离心5min后收集第三菌体沉淀;加20μl1mol/lsorbitol重悬第三菌体沉淀(质粒低于30kbp时可涡旋)后,添加3μl10mg/mlrnasea、420μllysisbuffer(0.05mtris-hcl,0.02medta,1%sds,ph12.8)混匀,放置于37℃培养30分钟中得到第四菌体溶液,每间隔10min上下颠倒混匀一次;加入420μldna抽提液(酚氯仿)混匀,放置于4℃离心机中,4200g离心30min;离心结束后,弃上清,用210μl70%乙醇溶液洗涤沉淀,放置于4℃离心机中,4200g离心5min;离心结束后,弃上清,可放置于65℃烘箱,干燥5min,或置于真空浓缩仪中45℃10min;干燥后,用20μlddh2o溶解dna。
将5μl第二质粒前体溶液转化至到大肠杆菌感受态细胞中,涂布到含有氨苄抗生素的lb平板,于30℃培养18小时,对转化子进行菌落pcr验证,挑选鉴定正确的菌落接种于含有氨苄抗生素的lb液体培养基,于30℃培养12-16小时后,利用商业常规质粒提取试剂盒将第二质粒前体进行提取并进行酶切验证及测序鉴定,富集得到纯度高、浓度大的第二质粒前体,其中,第二质粒前体包括标记基因b和第二限制性内切酶识别位点。
(3)①合成带有同源臂的供体序列:并添加待修改区域的两侧同源臂(45-500bp),生成供体序列,并发送基因合成公司合成带有同源臂的供体序列,要求将带有同源臂的供体序列克隆在细菌载体上(如puc57),并在合成片段的两端添加noti酶切位点,获得合成片段载体sg016,片段大小为4292bp,以及合成片段载体sg017,片段大小为3292bp。
②将供体序列和第二质粒前体分别进行线性化处理得到线性化供体序列和线性化第二质粒前体:利用新酶切位点srfi对第二质粒前体进行酶切,释放出线性化的第二质粒载体;利用noti对获得的合成片段载体sg016和sg017进行体外酶切,释放得到线性化的sg016和线性化的sg017,其中,对应酶切产物不需要进行胶回收直接就可以进行下一步修复。
(4)①将线性化供体序列和线性化第二质粒前体共转化至酿酒酵母细胞中:将线性化供体序列和线性化第二质粒前体的添加摩尔比为3:1,转化200-500ng有效dna(仅指接收载体和合成片段部分的总和)到酵母菌by4742中。其中,质粒前体释放的接收载体的实际用量为134ng,sg016合成片段部分用量为80ng,sg017合成片段部分用量为62ng;将线性化供体序列和线性化第二质粒前体共转化至酿酒酵母细胞中的过程如上述(2)③。
②筛选得到含有供体序列质粒的目的酿酒酵母细胞:将转化了供体序列和第二质粒前体的酿酒酵母细胞涂布于sc-his平板,于30℃条件下过夜培养得到第一平板菌落;将第一平板菌落影印至sc-his+5-foa(1g/l)平板、sc-ura及sc-his平板上,于30℃条件48小时,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定,筛选得到含有供体序列质粒的目的酿酒酵母细胞,其中,含有sg016合成片段的供体序列质粒命名为s4-v1,含有sg017合成片段的供体序列质粒命名为s4-v2。
其中,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定的步骤中,采用菌落pcr鉴定引物jso1279和菌落pcr鉴定引物jso1521分别对s4-v1和s4-v2进行鉴定,菌落pcr鉴定引物jso1279的序列如seq.idno.15,seq.idno.15如下所示:agtaggtggaatagctccagctatc;菌落pcr鉴定引物jso1521的序列如seq.idno.14,seq.idno.14如下所示:ctgaagttgcaagacaatctgaattg。
进行菌落pcr鉴定的菌落pcr反应体系如下:
挑取待检测菌落到20μl20mmnaoh中,置于pcr仪中,(98℃3min,4℃2min),重复5个循环,后静置备用;
再按照下表5的菌落pcr反应体系进行配制混合溶液,按照下表6所示的pcr反应程序进行pcr反应。
表5
表6
③富集含有供体序列质粒:将含有供体序列质粒从验证正确的酵母菌株中提取出来,具体过程如(2)⑤中步骤所示,将供体序列质粒转化至大肠杆菌感受态细胞中进行大量富集,利用商业常规质粒提取试剂盒将供体序列质粒进行提取并进行酶切验证及测序鉴定,富集得到纯度高、浓度大的供体序列质粒,有利于进行后续生物学相关研究。
结果分析:
实施例2的结果分析如下:
结果分析(一)
根据实施例2步骤(2)④中,将第三混合细胞溶液涂布到sc-ura的平板上,放置于30℃培养箱中培养48小时,并挑取5个菌落进行菌落pcr验证,菌落pcr验证结果如图10所示,采用筛选引物jso1221和筛选引物jso1521进行验证的pcr结果,可以发现5个菌落均有阳性条带,片段大小为871bp,为阳性克隆子,均为含有第二质粒前体的酿酒酵母细胞;采用物jso1061和引物12311进行验证时,得到的片段大小为586bp,由于改除序列比较特殊,扩增部分具有高gc%含量,导致利用常规的dna聚合酶及扩增条件,不能将其扩增出来,所以是没有条带。
进一步进行阳性率统计,如下表7所示,转化子为155个,阳性克隆比为5/5,阳性率为100%。
表7
结果分析(二):
根据实施例2步骤(3)①中,合成片段载体sg016和sg017如图11所示,图11为提供的待修改区域s4、标记基因ura3片段、供体序列sg016、供体序列sg017、修改后的s4-v1(含有sg016的供体序列质粒)、修改后的s4-v2(含有sg017的供体序列质粒)的序列比对结果。
结果分析(三)
根据实施例2步骤(3)②中,将供体序列和第二质粒前体分别进行线性化处理得到线性化供体序列和线性化第二质粒前体的步骤中,线性化结果如图12所示,lane1/2为第二质粒前体经srfi处理后的线性化第二质粒前体,片段大小为21457bp,lane3/4为合成的供体序列sg016质粒经noti处理后的线性化sg016,片段大小为4292bp,lane5/6为合成的供体序列sg017质粒经noti处理后的线性化sg017,片段大小为3292bp,均线性化完全,且浓度较高、无杂质。
结果分析(四):
根据实施例2步骤(4)②中,将第一平板菌落未生长的菌落影印至sc-his+5-foa(1g/l)平板及sc-ura平板上,于30℃条件48小时,选取sc-his+5-foa平板上的酵母菌落进行菌落pcr鉴定,筛选得到含有供体序列质粒的目的酿酒酵母细胞的步骤中,其中,sc-his+5-foa平板是为了在筛选正确组装的细胞的同时,除去因酶切处理不彻底而导致体内残留的质粒前体的假阳性酵母菌;sc-ura平板主要用于推断质粒前体残留的大致比例。选取sc-his+5-foa平板上的酵母菌落进行pcr鉴定,s4-v1菌落pcr鉴定结果如图13所示,阳性克隆子的片段大小为1050bp,s4-v2菌落pcr鉴定结果如图14所示,阳性克隆子的片段大小为950bp,发现s4-v1与s4-v2均有60%的酵母菌实现了正确组装修复,说明实施例1步骤(3)②的酶切产物直接进行转化组装,其效率很高,且不需要进行相应dna片段的胶回收纯化,过程简便快捷。进一步分析s4-v1与s4-v2的阳性率,如下表8所示:对于s4-v1片段,在sc-his+5-foa平板的转化子非常多,其中阳性克隆比为24/32,阳性率为75%;对于s4-v2片段,在sc-his+5-foa平板的转化子非常多,其中阳性克隆比为20/32,阳性率为62.5%。
表8
通过实施例2证明本发明提供的分子克隆方法将原始质粒大小为27.2kbp的质粒pjs380进行了多版本深度改造,实现了多个不连续区域的复杂修改,成功获得了无缝改造的s4-v1供体序列质粒和s4-v2供体序列质粒,该分子改造的方法操作十分简单,构建效率较高,构建成功率较高,适用于多版本、高通量的无缝克隆操作。
以上所述仅为本申请的较佳实施例而已,并不用以限制本申请,凡在本申请的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本申请的保护范围之内。
sequencelisting
<110>先进技术研究院
<120>基于合成基因和酿酒酵母同源重组机制的分子克隆方法
<130>2020-08-07
<160>15
<170>patentinversion3.3
<210>1
<211>77
<212>dna
<213>人工合成
<400>1
aaggattgccacatgttatatattgccgattatggcgctggcctgatcttcacaggcaga60
ttgtactgagagtgcac77
<210>2
<211>79
<212>dna
<213>人工合成
<400>2
gtggtaagggtttacacatacttcatcctttttaagattaaaagcatattcgcagttttc60
tgtgcggtatttcacaccg79
<210>3
<211>1276
<212>dna
<213>人工合成
<400>3
aaggattgccacatgttatatattgccgattatggcgctggcctgatcttcacaggcaga60
ttgtactgagagtgcaccataccacagcttttcaattcaattcatcattttttttttatt120
cttttttttgatttcggtttctttgaaatttttttgattcggtaatctccgaacagaagg180
aagaacgaaggaaggagcacagacttagattggtatatatacgcatatgtagtgttgaag240
aaacatgaaattgcccagtattcttaacccaactgcacagaacaaaaacctgcaggaaac300
gaagataaatcatgtcgaaagctacatataaggaacgtgctgctactcatcctagtcctg360
ttgctgccaagctatttaatatcatgcacgaaaagcaaacaaacttgtgtgcttcattgg420
atgttcgtaccaccaaggaattactggagttagttgaagcattaggtcccaaaatttgtt480
tactaaaaacacatgtggatatcttgactgatttttccatggagggcacagttaagccgc540
taaaggcattatccgccaagtacaattttttactcttcgaagacagaaaatttgctgaca600
ttggtaatacagtcaaattgcagtactctgcgggtgtatacagaatagcagaatgggcag660
acattacgaatgcacacggtgtggtgggcccaggtattgttagcggtttgaagcaggcgg720
cagaagaagtaacaaaggaacctagaggccttttgatgttagcagaattgtcatgcaagg780
gctccctatctactggagaatatactaagggtactgttgacattgcgaagagcgacaaag840
attttgttatcggctttattgctcaaagagacatgggtggaagagatgaaggttacgatt900
ggttgattatgacacccggtgtgggtttagatgacaagggagacgcattgggtcaacagt960
atagaaccgtggatgatgtggtctctacaggatctgacattattattgttggaagaggac1020
tatttgcaaagggaagggatgctaaggtagagggtgaacgttacagaaaagcaggctggg1080
aagcatatttgagaagatgcggccagcaaaactaaaaaactgtattataagtaaatgcat1140
gtatactaaactcacaaattagagcttcaatttaattatatcagttattaccctatgcgg1200
tgtgaaataccgcacagaaaactgcgaatatgcttttaatcttaaaaaggatgaagtatg1260
tgtaaacccttaccac1276
<210>4
<211>22
<212>dna
<213>人工合成
<400>4
cccaacaggtctcttgatggtc22
<210>5
<211>24
<212>dna
<213>人工合成
<400>5
cgtggatgatgtggtctctacagg24
<210>6
<211>20
<212>dna
<213>人工合成
<400>6
ctgtgctccttccttcgttc20
<210>7
<211>82
<212>dna
<213>人工合成
<400>7
tcgaaaaggattgccacatgttatatattgccgattatggcgctggcctgatcttcacag60
tcaccacgagctgaaagctatc82
<210>8
<211>82
<212>dna
<213>人工合成
<400>8
agtggtaagggtttacacatacttcatcctttttaagattaaaagcatattcgcagtttt60
cgatagctttcagctcgtggtg82
<210>9
<211>26
<212>dna
<213>人工合成
<400>9
cttgcaatattcctacctggtgtctc26
<210>10
<211>84
<212>dna
<213>人工合成
<400>10
caccgcccgagcccaggtaaccgcgccatgtcccctccccttcccccggccgggcccggg60
cgcagattgtactgagagtgcacc84
<210>11
<211>85
<212>dna
<213>人工合成
<400>11
ttgtattttgtagtccaccatcctgataaggttaagggccccaacggtaaaagaccgccc60
gggctctgtgcggtatttcacaccg85
<210>12
<211>1288
<212>dna
<213>人工合成
<400>12
ttgtattttgtagtccaccatcctgataaggttaagggccccaacggtaaaagaccgccc60
gggctctgtgcggtatttcacaccgcatagggtaataactgatataattaaattgaagct120
ctaatttgtgagtttagtatacatgcatttacttataatacagttttttagttttgctgg180
ccgcatcttctcaaatatgcttcccagcctgcttttctgtaacgttcaccctctacctta240
gcatcccttccctttgcaaatagtcctcttccaacaataataatgtcagatcctgtagag300
accacatcatccacggttctatactgttgacccaatgcgtctcccttgtcatctaaaccc360
acaccgggtgtcataatcaaccaatcgtaaccttcatctcttccacccatgtctctttga420
gcaataaagccgataacaaaatctttgtcgctcttcgcaatgtcaacagtacccttagta480
tattctccagtagatagggagcccttgcatgacaattctgctaacatcaaaaggcctcta540
ggttcctttgttacttcttctgccgcctgcttcaaaccgctaacaatacctgggcccacc600
acaccgtgtgcattcgtaatgtctgcccattctgctattctgtatacacccgcagagtac660
tgcaatttgactgtattaccaatgtcagcaaattttctgtcttcgaagagtaaaaaattg720
tacttggcggataatgcctttagcggcttaactgtgccctccatggaaaaatcagtcaag780
atatccacatgtgtttttagtaaacaaattttgggacctaatgcttcaactaactccagt840
aattccttggtggtacgaacatccaatgaagcacacaagtttgtttgcttttcgtgcatg900
atattaaatagcttggcagcaacaggactaggatgagtagcagcacgttccttatatgta960
gctttcgacatgatttatcttcgtttcctgcaggtttttgttctgtgcagttgggttaag1020
aatactgggcaatttcatgtttcttcaacactacatatgcgtatatataccaatctaagt1080
ctgtgctccttccttcgttcttccttctgttcggagattaccgaatcaaaaaaatttcaa1140
agaaaccgaaatcaaaaaaaagaataaaaaaaaaatgatgaattgaattgaaaagctgtg1200
gtatggtgcactctcagtacaatctgcgcccgggcccggccgggggaaggggaggggaca1260
tggcgcggttacctgggctcgggcggtg1288
<210>13
<211>22
<212>dna
<213>人工合成
<400>13
gacaagttggcagcaacaacac22
<210>14
<211>26
<212>dna
<213>人工合成
<400>14
ctgaagttgcaagacaatctgaattg26
<210>15
<211>25
<212>dna
<213>人工合成
<400>15
agtaggtggaatagctccagctatc25
- 还没有人留言评论。精彩留言会获得点赞!