帕拉米松乙酸酯及帕拉米松的制备方法与流程
1.本发明属于有机合成及药物化学技术领域,具体涉及一种帕拉米松乙酸酯及帕拉米松的制备方法。
背景技术:
2.帕拉米松乙酸酯(paramethasone acetate),化学名称为21-乙酰氧基-6α-氟-11β,17-二羟基-16α-甲基-1,4-二烯-3,20-二酮,是一种糖皮质激素,由lilly公司开发,该产品于1961年4月17日由美国食品药品监督管理局(fda)批准上市,其商品名称为haldrone,主要用于过敏性与炎症性疾病。
3.帕拉米松(paramethasone),化学名称为6α-氟-11β,17,21-三羟基-16α-甲基-1,4-孕二烯-3,20-二酮,主要用于严重的细菌感染和严重的过敏性疾病、各种各种血小板减少性紫癜、粒细胞减少症、严重皮肤病、器官移植的免疫排斥反应、肿瘤的治疗及对糖皮质激素敏感的眼部炎症等,抗炎活性相当于泼尼松龙的2.5倍,无水钠潴留作用。
4.传统的帕拉米松乙酸酯的制备方法,是以d jerassi等报道的工艺为参考进行的,具体采用醋酸妊娠双烯醇酮为起始物料、经过11步反应,才能合成获得帕拉米松乙酸酯,其工艺路线如下:
[0005][0006]
该工艺具体可参照文献djerassi,c.et al.steroids.cxxxvi.synthesis of a new class of potent cortical hormones 6a-fluoro-6a,9 a-difluoro-16a-methylprednisolone and related steroids.:j.am.chem.soc.(jacsat)82,2318(1960)的内容。
[0007]
现有技术采用醋酸妊娠双烯醇酮为起始物料,该材料提取自植物黄姜,而黄姜产量受天气影响很大,国内的黄姜产量不够稳定,原料价格也经常大幅变动,不利于控制生产成本。
[0008]
此外现有工艺的路线较长,副产物较多,对原料的利用率不高,最终产品的收率比较低。
技术实现要素:
[0009]
针对上述问题,本发明提供一种帕拉米松乙酸酯及帕拉米松的制备方法。
[0010]
一方面,本发明提供一种帕拉米松乙酸酯的制备方法,采用如下技术方案:
[0011]
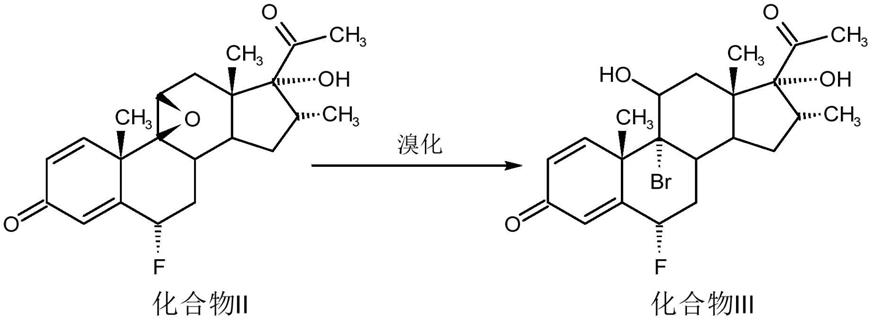
以下式所示的化合物i为原料,化合物i为16-甲基羟基环氧黄体酮,依次经过氟化、溴化、脱溴、碘化和置换反应得到化合物vi,化合物vi为帕拉米松乙酸酯,反应式如下:
[0012][0013]
其中,反应步骤如下:
[0014]
s1氟化反应:
[0015][0016]
在氮气保护下,取1质量份数的化合物i加入5体积份数的第一有机溶剂中,加入苯甲酰氯在40-85℃条件下反应2-8小时后,再加入0.5-2体积份数的甲醇反应,继续降温至-20-0℃,加入1-5体积份数的乙腈、0.2-2体积份数的水后,加入选择性氟反应至原料反应完全,在盐水体系中结晶、过滤、干燥后得到化合物ii;
[0017]
s2溴化反应:
[0018][0019]
取1质量份数的化合物ii溶于1-10体积份数丙酮溶剂中,滴加氢溴酸至反应完全,将产物洗出,过滤、干燥后得化合物iii;
[0020]
s3脱溴反应:
[0021][0022]
取1质量份数的化合物iii溶于1-10体积份数的第二有机溶剂中,加入0.5体积份数的甲基含氢硅油、0.2-3体积份数的三丁基氧化锡和0.005-0.05质量份数的偶氮二异丁腈,在50-75℃回流反应2-6h,反应完全后蒸馏至固体析出,加入第三有机溶剂置换出第二有机溶剂,经蒸馏、结晶、干燥后得化合物iv;
[0023]
s4碘化反应:
[0024][0025]
取1质量份数的化合物iv溶于1-10体积份数的低级脂肪醇溶剂中,依次加入适量氯化钙和氧化钙,在低温条件下滴加碘液至反应完全,再加入第四有机溶剂,结晶、过滤、洗涤、干燥后得化合物v;
[0026]
s5置换反应:
[0027][0028]
取1质量份数的化合物v溶于1-10体积份数的丙酮溶剂中,加入乙酸钾、冰乙酸和
水,在40-80℃条件下反应2-8小时,反应完全后洗涤,加入脱色剂,过滤、结晶、二次过滤、干燥后得到化合物vi,即帕拉米松乙酸酯。
[0029]
进一步优选的,
[0030]
步骤s1中,第一有机溶剂为乙腈、吡啶或两者的混合;盐水体系为亚硫酸盐、偏重亚硫酸盐或两者的混合。
[0031]
步骤s3中,第二有机溶剂为四氢呋喃、2-甲基四氢呋喃、乙醇、异丙醇、丙酮、丁酮、二氯甲烷、n,n-二甲基甲酰胺、乙腈、乙酸乙酯中的一种或多种;第三有机溶剂为乙酸乙酯。
[0032]
步骤s4中,低级脂肪醇溶剂为甲醇或乙醇溶液;第四有机溶剂为乙酸、羟基乙酸、柠檬酸、苯甲酸中的一种或多种。
[0033]
步骤s5中,脱色剂为活性炭、氧化铝、活性白土、硅藻土中的一种或多种。
[0034]
另一方面,本发明提供一种利用帕拉米松乙酸酯水解获取帕拉米松的制备方法,其使用的帕拉米松乙酸酯为采用前述制备方法获得的化合物vi,具体步骤为:取1体积份数的化合物vi酯化物溶液,在氮气保护下,加入1-10体积份数的醇类溶液及碱溶液,在-20-0℃的条件下反应至原料反应完全,中和、蒸馏后加入置换剂置换醇类溶剂,二次蒸馏至固体析出,结晶、过滤、洗涤后得到化合物vii,即帕拉米松,反应式如下:
[0035][0036]
优选的,醇类溶液为甲醇溶液,碱溶液为氢氧化钾溶液,置换剂为异丙醚。
[0037]
本发明的有益效果是:
[0038]
1、采用16-甲基羟基环氧黄体酮为起始物料,该物料来源于微生物发酵,产品不受自然因素影响,产能有保障,物料价格平稳,利于控制、降低生产成本;
[0039]
2、反应路线短,产品收率大幅提高,副产物产生少;经测试本制备方法的产品纯度达到99.2%以上,总重量收率可达到55%以上,具有很好的应用前景。
具体实施方式
[0040]
下面通过实施方式对本发明进行进一步详细的说明。
[0041]
实施例1化合物ii的制备
[0042]
向反应瓶中加入1质量份数的化合物ⅰ(16-甲基羟基环氧黄体酮)、3体积份数的乙睛和2体积份数的吡啶,通氮气排空气2次,室温下搅拌10-15min,然后加入0.5体积份数的苯甲酰氯,搅拌下缓慢升温到85℃反应4小时,tlc监测反应。反应液冷水浴降温至40℃,然后向反应液中滴加1.0体积份数的甲醇,滴加时间10-15min,然后继续在40℃下反应30min,反应掉过量的苯甲酰氯,然后降温至﹣10℃,向反应液中加入1体积份数的乙睛和0.5体积份
数的水,搅拌10-15min,分次将选择性氟加入到上述反应液中,反应3h,直至有大量淡黄色固体析出。另外使用0.01质量份数的偏重亚硫酸钠和8体积份数的水配制好预冷溶液,预冷至0-5℃,向反应液中预冷液,温度保持在0-5℃,滴加55min,继续搅拌2h,静止2h析出固体,过滤,滤饼用甲醇洗涤,在60-65℃下将固体烘干24h,得到淡黄色氟化物固体,即化合物ii,重量收率92.12%,产品纯度94.55%,其液相色谱数据如下表:
[0043]
表1.化合物ii的液相色谱数据
[0044][0045][0046]
实施例2化合物iii的制备
[0047]
向反应瓶中加入1质量份数的化合物ii和8体积份数的丙酮,搅拌10-15min,冷却至0-5℃,滴加3.5体积份数的氢溴酸溶液,滴加时间30-40min,继续反应4.5-h至溶解清亮,tlc监测反应。反应完全后,滴入预冷至0-5℃的30体积份数的水,滴加时间40-60min,滴加时温度条件为0-10℃,搅拌1-1.5h,析出固体,过滤,用水洗至中性,固体在40-50℃下烘干16h,得到淡黄色上溴物固体,即化合物iii,重量收率107%,产品纯度94.35%,其液相色谱数据如下表:
[0048]
表2.化合物iii的液相色谱数据
[0049][0050][0051]
实施例3化合物iv的制备
[0052]
向反应瓶中加入1质量份数的化合物iii、10体积份数的四氢呋喃、0.5体积份数的三丁基氧化锡和0.5体积份数的甲基含氢硅油,通氮气排空气2-3次,然后加入0.02质量份数的偶氮二异丁腈,升温至60-65℃回流反应4-6h,tlc监测反应。反应完全后,冷却至20-40℃,减压蒸馏至有大量固体析出时,析出约4-5体积份数时,加入2体积份数的乙酸乙酯,继续蒸馏,重复3次。冷却至﹣5-0℃,3-4h冷却结晶。过滤,用预冷异丙醚洗涤,固体在60-65℃下烘干24h,得到微黄色脱溴物固体,即化合物iv,重量收率76.80%,产品纯度96.68%,其液相色谱数据如下表:
[0053]
表3.化合物iv的液相色谱数据
[0054]
峰保留时间面积面积%峰宽峰高13.39013.897570.14990.19971.0974823.96576.140400.82130.17126.9860935.78955.443480.59810.29082.7317147.48868.313060.73690.23264.6168658.0068963.1796996.68420.2731509.8242269.20126.348080.28420.34391.07512711.70237.809850.40780.36311.54717813.53729.443090.31760.35231.07255总计 9270.57522100
ꢀꢀ
[0055]
实施例4化合物v的制备
[0056]
向反应瓶中加入6体积份数甲醇和0.4质量份数氯化钙,室温下搅拌溶解,加入1质量份数化合物iv,搅拌10-15min,然后加入0.6质量份数氧化钙,边搅拌边降温,降温至0-5℃,避光存储。向另一反应瓶中加入4体积份数甲醇和0.4质量份数氯化钙,搅拌溶解,加入1.2质量份数的碘,继续搅拌溶解获得碘液。向溶有化合物iv的反应液中滴加碘液,滴加时温度条件设置为0-5℃,滴加时间2-4h,反应0.5-4h,tlc监测反应。将反应液缓缓加入由10
体积份数水和1.15体积份数冰乙酸混合得到的预冷液中,调节温度至0-10℃,搅拌2-6h结晶,过滤,用少量预冷水洗涤,得到淡黄色固体,在30-35℃下真空干燥24h,得到上碘物,即化合物v。
[0057]
实施例5化合物vi(帕拉米松乙酸酯)的制备
[0058]
向反应瓶中加入15体积份数丙酮、4质量份数乙酸钾、0.3体积份数冰乙酸和0.2质量份数的水,通氮气置换3次,加入1质量份数化合物v,升温至56-60℃回流反应2-8h,tlc监测反应,反应完成后,加入饱和氯化钠溶液洗涤,将反应液加入中性氧化铝,在40℃条件下脱色30分钟,过滤得到酯化物溶液,酯化物溶液浓缩后,留少量母液,冷冻出料,过滤,烘干得化合物vi(帕拉米松乙酸酯),产品纯度98.22%,其液相色谱数据如下表:
[0059]
表4.化合物vi的液相色谱数据
[0060]
峰保留时间面积面积%峰宽峰高13.93122.045850.25760.13242.5625624.4776.899520.08060.26590.35635035.71828.599510.33420.22211.8526047.36120.112110.23500.24821.2208457.8868406.0439598.22180.2269573.9975069.08513.283470.15520.28160.587944710.2848.378300.09790.26320.458384811.45213.723010.16030.30220.689436913.21139.139880.45730.35181.74402总计 8558.22561100
ꢀꢀ
[0061]
实施例6化合物vii(帕拉米松)的制备
[0062]
取实施例5制得的化合物vi溶液,在氮气保护下,搅拌10-15min,冷却至﹣10-﹣5℃,加入10体积份数甲醇和0.2质量份数的氢氧化钾,搅拌溶解,再向反应液中滴加30%氢氧化钾/甲醇溶液,滴加温度﹣10-﹣5℃,滴加时间0.5-3h,反应30-50min,反应液逐渐变清亮。tlc监测反应,反应完全后,向反应液中加入0.1体积份数冰乙酸中和调节ph至6.0-8.0,减压浓缩至3-4体积份数,加入3体积份数的异丙醚,继续蒸馏至3-4体积份数,重复3次,直至有大量白色固体析出,冷却至0-5℃静置2-3h,过滤,先用1体积份数的预冷水洗涤,再用预冷的异丙醚洗涤,在50-60℃下烘干24h,得固体粗品,再加入15体积份数甲醇加热溶解,再次过滤、浓缩,冷却并过滤,烘干得到化合物vii,即帕拉美松,以化合物v为起始物料计,重量收率83.50%,产品纯度99.22%,其液相色谱数据如下表:
[0063]
表5.化合物vii的液相色谱数据
[0064]
峰保留时间面积面积%峰宽峰高10.8661.144890.01460.19250.076536423.1533.595180.04590.12560.42535133.3807.712310.09840.13730.80866844.8704.479710.05720.26970.22075455.7325.190650.06630.18830.39755867.2406.544340.08350.24720.373964
79.5303.385940.04320.19300.254790810.5727773.4897599.22190.3060368.71204913.42515.530780.19820.44470.4836231014.4513.744870.04780.39920.1128471121.1613.098560.03960.34890.1147871224.9772.644050.03370.42060.07549921326.1793.891040.04970.45820.102191总计 7834.45206100
ꢀꢀ
[0065]
上述实施例中,质量份数与体积份数的比的单位为g/ml。
[0066]
根据上述实施例数据可知,本发明提供的制备工艺,经过六步反应和得到帕美松成品,在不考虑母液回收套用的情况下,总重量收率可达55%以上。
[0067]
综上,本发明提供了一种帕拉米松乙酸酯及帕拉米松的制备方法,以微生物发酵获得的化合物ⅰ为起始原料,避免植物来源的限制;通过6步反应合成帕拉米松,工艺路线大大缩短,大幅提高产品收率,且不涉及危险化学工艺,绿色清洁,大大提高原子经济性;化合物ⅱ的制备中采用了偏重亚硫酸钠,用于产品精制结晶,大幅提高了产品的收率和纯度,收率提高5%以上;化合物ⅳ的制备过程中,采用有机溶剂异丙醚对产品进行洗涤,有效降低了产品杂质,纯度由90%提升至93-96%;化合物
ⅴ
的制备过程中采用乙酸溶液体系进行精制,也有效提高产物收率;化合物ⅵ的制备过程中采用饱和氯化钠溶液洗和中性氧化铝脱色,产品外观得到较大改善,同时有效提高收率和质量。此外本合成方法工艺操作简便,易于放大,在实际的工业生产中,通过母液回收套用,可进一步提升收率,应用前景良好。
[0068]
本领域的技术人员可以明确,在不脱离本发明的总体精神以及构思的情形下,可以做出对于以上实施例的各种变型。其均落入本发明的保护范围之内。本发明的保护方案以本发明所附的权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1