一种抗新冠药物Molnupiravir的制备方法与流程
一种抗新冠药物molnupiravir的制备方法
技术领域
1.本发明属于药物合成化学领域,具体涉及一种抗新冠药物molnupiravir的制备方法。
背景技术:
2.molnupiravir(又称mk4482或eidd2801)是埃默里大学开发的一款具有广谱抗rna病毒活性的核糖核苷类类似物,可有效抑制流感病毒、丙型肝炎病毒、埃博拉病毒、呼吸道合胞病毒、冠状病毒如sars-cov-1和mers病毒等的复制。埃默里大学将eidd2801授权给默沙东和 ridgeback,由二者共同开发成为口服抗新型冠状病毒候选药物。临床试验结果显示,molnupiravir可将新冠患者住院或死亡风险降低30%。2021年12月23 日,fda授予molnupiravir紧急使用授权(eua),用于治疗新型冠状病毒 (sars-cov-2)检测阳性,且有较高风险发展为重症的轻症或中症新冠肺炎 (covid-19)的成人患者。
3.molnupiravir的化学名称为尿苷5'-甲基丙酸酯-4-肟,其化学结构如下所示:
[0004][0005]
专利wo2019113462首先公开了molnupiravir的合成路线。该路线采用尿苷为原料,首先对顺式邻二羟基进行保护,再与异丙酸酐反应生成异丁酸酯。中间体异丁酸酯在三氯氧磷作用下与1,2,4-三氮唑反应,生产三氮唑中间体;三氮唑中间体再与羟胺反应,最后脱去保护基得molnupiravir,总收率为17%。
[0006][0007]
synlett(2021,32,326-328)报道了一条以胞苷为起始物料合成molnupiravir 的
路线。首先用丙酮对胞苷的顺式邻二羟基进行保护,再与异丁酸酐反应生成异丁酸酯中间体;异丁酸酯中间体与硫酸羟胺反应,再脱除保护得molnupiravir,总收率为44%。
[0008][0009]
chem.commun.(2020,56,13363-13364)报道了molnupiravir的酶法合成路线。该路线同样以胞苷为起始物料,首先胞苷在诺维信脂肪酶435作用下进行选择性酯化,生成异丁酸酯中间体;中间体再与硫酸羟胺反应得molnupiravir,两步总收率为75%。
[0010][0011]
虽然molnupiravir的化学法或酶法合成取得了较大进展,但是目前化学法合成步骤较长,收率较低;酶法虽然步骤较短,收率高,但是酶用量大,价格昂贵,综合成本高。因此,开发一种路线简洁、工艺简单可靠、综合成本低且适合工业化生产的molnupiravir合成路线在目前显得尤为重要。
技术实现要素:
[0012]
本发明的目的在于提供一种工艺简单可靠、综合成本低且易于商业化生产的 molnupiravir(化合物i)制备方法。
[0013]
本发明的合成路线如下:
[0014][0015]
本发明包括以下步骤:
[0016]
1)尿苷(化合物ii)在路易斯酸作用下与异丁酸酐(化合物iii)进行酰化反应,得到化合物iv。
[0017]
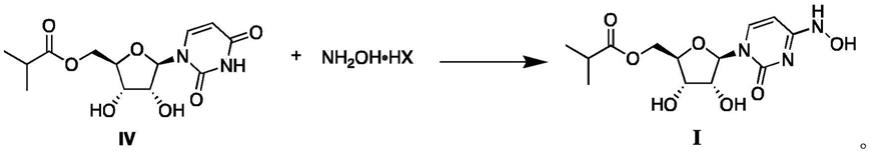
2)化合物iv在硅试剂存在下与羟胺盐酸盐或羟胺硫酸盐发生反应,得到化合物i。
[0018]
在步骤1)所述路易斯酸为三氟甲烷磺酸铜、三氟甲烷磺酸锌、三氟甲烷磺酸镍、三氟甲烷磺酸镧中的一种或多种。
[0019]
进一步地,在步骤1)中所述路易斯酸与化合物ii的摩尔比为1:20~1:1000。
[0020]
进一步地,在步骤1)中所述化合物ii与化合物iii的摩尔比为1:1~1:2。
[0021]
进一步地,在步骤1)中所述酰化反应的温度为-10℃~50℃。
[0022]
在步骤2)中所述硅试剂为六甲基二硅胺烷/三氟甲磺酸三甲基硅酯、六甲基二硅胺烷/三甲基氯硅烷中的一种或多种。
[0023]
进一步地,在步骤2)中所述硅试剂中的六甲基二硅胺烷与化合物iv的摩尔比为2:1~10:1;三氟甲磺酸三甲基硅酯或三甲基氯硅烷与化合物iv的摩尔比为0.01:1~0.2:1。
[0024]
进一步地,在步骤2)中所述化合物iv与盐酸羟胺或硫酸羟胺的摩尔比为 1:1~1:2。
[0025]
与现有技术/文献相比,本发明具有以下的显著优点:
[0026]
1)本发明采用路易斯酸催化剂催化尿苷进行选择性酰化,副产物少。
[0027]
2)本发明步骤短,三废量大大降低,整个制备过程更加绿色环保;
[0028]
3)本发明工艺简单可靠,综合生产成本低,因此具有良好的市场竞争力。
具体实施方式
[0029]
以下实施例向本领域普通技术人员提供如何制造和评价本发明,所述实施例仅是本公开内容的示范且不圈定限制范围。尽管已经尽力确保关于数值(例如,量、温度等)的准确性,但是应当考虑一些误差和偏差。除非另外说明,否则温度是以℃为单位或者在环境温度下,且压力是在大气压下或附近。
[0030]
本实施例中所叙述的用于本发明所描述的公开化合物的制备方法是众多方法中
的一种,本发明申请公开化合物的制备方法尚有很多其他方法,本技术不圈定限制范围。因此,本公开内容所属颁域的技术人员可容易地修改所叙述的方法或者利用不同的方法来制备所公开的化合物的一种或多种。下列方法仅是示例性的,温度、催化剂、浓度、反应物组成、以及其它工艺条件可改变,并且对于期望的化合物,本公开内容所属领域的技术人员可以容易的选择合适的反应物和条件进行制备。
[0031]
实施例1
[0032]
化合物iv的制备
[0033]
244.2g化合物ii和3.62g三氟甲磺酸铜溶解于2l的丁酮,室温下滴加166g异丁酸酐,搅拌至反应完全。减压除去丁酮,残余物加入1l乙酸乙酯和500ml水,萃取分液。水相再次用1l乙酸乙酯萃取。合并的有机相用10%碳酸氢钠洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/正庚烷重结晶,得到286g化合物iv,收率91%,hplc纯度99.3%。
[0034]1h nmr(dmso-d6,400mhz)δ11.38(s,1h),7.62(d,j=8.0hz,1h),5.75(d,j= 4.1hz,1h),5.66(d,j=7.8hz,1h),5.52(br s,1h),5.32(br s,1h),4.28-4.14(m, 2h),4.10-4.04(m,1h),4.02-3.92(m,2h),2.62-2.52(m,1h),1.09(d,j=6.8hz, 6h);
[0035]
13
c nmr(dmso-d6,101mhz)δ176.4,163.5,151.0,141.1,102.4,89.1,81.5,73.2, 70.1,64.1,33.6,19.2;
[0036]
hrms(esi):m/z calcd for c
13h18
n2o7[m+h]
+
315.1187,found:315.1182.
[0037]
实施例2
[0038]
化合物iv的制备
[0039]
244.2g化合物ii和3.64g三氟甲磺酸锌溶解于2l的丁酮,室温下滴加166g异丁酸酐,搅拌至反应完全。减压除去丁酮,残余物加入1l乙酸乙酯和500ml水,萃取分液。水相再次用1l乙酸乙酯萃取。合并的有机相用10%碳酸氢钠洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/正庚烷重结晶,得到274g化合物iv,收率87%,hplc纯度99.2%。
[0040]
实施例3
[0041]
化合物iv的制备
[0042]
244.2g化合物ii和3.57g三氟甲磺酸镍溶解于2l的丁酮,室温下滴加166g异丁酸酐,搅拌至反应完全。减压除去丁酮,残余物加入1l乙酸乙酯和500ml水,萃取分液。水相再次用1l乙酸乙酯萃取。合并的有机相用10%碳酸氢钠洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/正庚烷重结晶,得到226g化合物iv,收率72%,hplc纯度99.0%。
[0043]
实施例4
[0044]
化合物iv的制备
[0045]
244.2g化合物ii和5.86g三氟甲磺酸镧溶解于2l的丁酮,室温下滴加166g异丁酸酐,搅拌至反应完全。减压除去丁酮,残余物加入1l乙酸乙酯和500ml水,萃取分液。水相再次用1l乙酸乙酯萃取。合并的有机相用10%碳酸氢钠洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/正庚烷重结晶,得到289g化合物iv,收率92%,hplc纯度99.1%。
[0046]
实施例5
[0047]
化合物i的制备
[0048]
氮气氛下,向157g化合物iv和323g六甲基二硅氮烷悬浮液中加入5.5g三氟甲磺酸三甲基硅酯,加热(80℃)搅拌3小时。然后再加入100g硫酸羟胺,保温搅拌至反应完全,降
温,减压浓缩。残余物加入1l乙酸乙酯和500ml水,萃取分液。有机相加入10ml冰醋酸,室温搅拌2小时后用10%碳酸氢钠溶液洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/甲叔醚重结晶,得到147g化合物i,收率89%,hplc纯度99.8%。
[0049]1h nmr(dmso-d6,400mhz)δ10.02(s,1h),9.67(s,1h),6.82(d,j=8.2hz, 1h),5.71(d,j=5.5hz,1h),5.59(d,j=8.2hz,1h),5.38(d,j=5.5hz,1h),5.23 (d,j=4.5hz,1h),4.2(dd,j=12.0,3.1hz,1h),4.13(dd,j=12.0,4.9hz,1h), 4.00(q,j=5.3hz,1h),3.92(h,j=4.6hz,2h),2.57(p,j=7.0hz,1h),1.09(d,j =7.0hz,6h);
[0050]
13
c nmr(dmso-d6,101mhz)δ176.4,149.9,143.7,130.3,99.2,88.1,81.1,72.4, 70.4,64.3,33.6,19.2(4),19.2(2);
[0051]
hrms(esi):m/z calcd for c
13h19
n3o7[m+h]
+
330.1296,found:330.1294.
[0052]
实施例6
[0053]
化合物i的制备
[0054]
氮气氛下,向157g化合物iv和400g六甲基二硅氮烷悬浮液中加入10g三甲基氯硅烷,加热(80℃)搅拌3小时。然后再加入100g硫酸羟胺,保温搅拌至反应完全,降温,减压浓缩。残余物加入1l乙酸乙酯和500ml水,萃取分液。有机相加入10ml冰醋酸,室温搅拌2小时后用10%碳酸氢钠溶液洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/甲叔醚重结晶,得到134g化合物i,收率81%, hplc纯度99.7%。
[0055]
实施例7
[0056]
化合物i的制备
[0057]
氮气氛下,向157g化合物iv和500g六甲基二硅氮烷悬浮液中加入10g三氟甲磺酸三甲基硅酯,加热(80℃)搅拌3小时。然后再加入42g盐酸羟胺,保温搅拌至反应完全,降温,减压浓缩。残余物加入2l乙酸乙酯和500ml水,萃取分液。有机相加入10ml冰醋酸,室温搅拌2小时后用10%碳酸氢钠溶液洗涤,无水硫酸镁干燥,减压浓干。得到的粗品经乙酸乙酯/甲叔醚重结晶,得到152g化合物i,收率92%,hplc纯度99.7%。
[0058]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1