一种负性液晶化合物及其制备方法与流程
本发明属于液晶化合物
技术领域:
,具体地说,涉及一种负性液晶化合物及其制备方法。
背景技术:
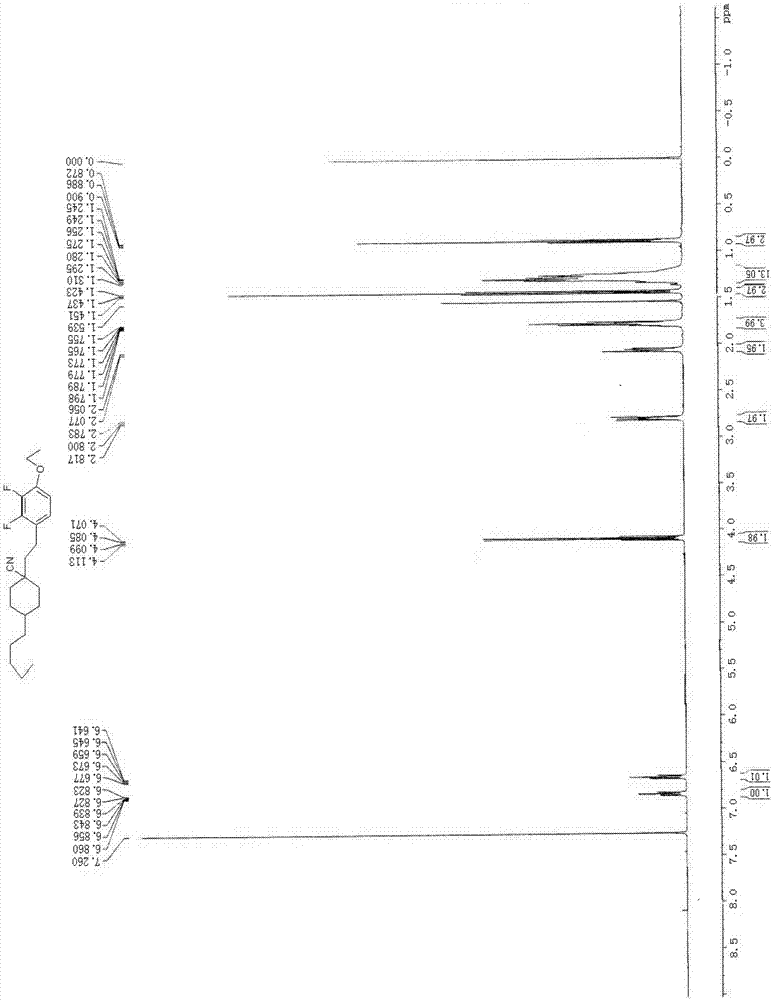
:近十几年液晶显示技术得到了迅速的发展,液晶显示产品在人们普通生活得到了快速普及。新型液晶显示方式主要有光学补偿弯曲模式(ocb)、共面转变液晶显示(ips)、垂直取向模式(va)、轴对称微结构液晶显示(asm)、多畴扭曲液晶显示等。各种显示方式液晶盒的设计不同、驱动方式不同,液晶分子指向矢和玻璃基板方向不同,光学补偿弯曲模式(ocb)、共面转变液晶显示(ips)液晶分子指向矢和玻璃基板方向是平行的,而垂直取向模式(va)、轴对称微结构液晶显示(asm)在无电场状态下液晶分子指向矢和玻璃基板方向垂直。平行排列方式的ips,液晶的介电各向异性(δε)既可以是正的,也可以是负的。垂直取向模式(va)所有液晶分子在零场时和玻璃基板方向垂直,与垂直入射光线平行。当偏振片正交时,会显示良好的暗态,所以该类器件有良好的对比度,用到液晶的介电各向异性(δε)必须是负的。液晶的光学各向异性(δη)、液晶盒的厚度(d)、入射光的波长(λ)几乎影响不到对比度。垂直取向模式(va)的响应时间比扭曲型器件要短得多,约为一半左右。在外加电压影响下,va器件主要产生的是液晶分子的弯曲形变,ecb产生的是液晶分子的展曲形变,而扭曲显示产生的是液晶分子的扭曲形变,其响应时间也分别与弯曲、展曲、扭曲弹性常数成反比,由于大部分的液晶在通常的情况下液晶的弯曲弹性常数大于展曲弹性常数,展曲弹性常数又大于扭曲弹性常数,这也是va器件响应时间较快的原因。为了能使显示器件的性能更加接近理想化,人们一直在致力于研究新的液晶化合物,这得以使液晶化合物及显示器件的性能仍在不断的向前发展。近几年,侧位含氟,含氰等多种负性材料在液晶混合物中得到了大量应用。在文献mol.cryst.liq.cryst.,1983,vol.94,pp109-118提到侧位含二氰基的液晶酯类化合物,如该类化合物具有大的负介电各向异性,但也具有粘度大、电阻率低、稳定性差和混溶性差等缺点,限制了其应用范围。在us5279764、us2011309300等专利中提到了众多侧位二氟液晶化合物,例如此类液晶化合物具有负介电各向异性适中,粘度低,电阻率高以及稳定性好等特点,在各种显示模式中具有广泛应用。在us4510069(1985年)专利中提到多种烷基取代的环己基腈衍生物。这一类液晶化合物具有较强的负介电各向异性,稳定性,清亮点适中。例如有鉴于此特提出本发明。技术实现要素:本发明要解决的技术问题在于克服现有技术的不足,提供一种新的负性液晶化合物及其制备方法。为实现本发明的目的,本发明采用如下技术方案:一种负性液晶化合物,结构通式如式(i)所示:其中:r1、r2均独立选自以下基团中的任意一种:h或碳原子1-10的烷基或烷氧基;或碳原子2-10的直链烯基或烯氧基;或碳原子1-10的氟代直链烷基,或碳原子2-10的氟代直链烯烃基,或-cl,-f,-ocf3,-ocf2h;或含烷基取代基或氟取代基的芳烃基;或含有或无取代基的环丙基或环丁基或环戊基;或其他含取代基的含氧或氮的五元或六元杂环基;或碳原子1-20的烷基酰氧基或芳基酰氧基;n=1或2;x、y均独立选自h或f;选自以下基团的任意一种:优选,本发明所述的负性液晶化合物,其中,n=1,x、y均为h。本发明所述的负性液晶化合物优选下述式(i-1)至式(i-7)中的任意一种:本发明所述的负性液晶化合物更优选下述式(i-8)至式(i-11)中的任意一种:现有技术中的液晶化合物,如负介电各向异性强,但稳定性很差,混溶性差,限制了其应用。本发明的负性液晶化合物借鉴了这两类液晶化合物产品的结构优点,稳定性、粘度和混溶性相似,而相对于这两类产品,在负介电各向异性性能上得到明显提升,可降低混晶产品中驱动电压,化学稳定性好,混溶性好。本发明的目的还在于提供所述的负性液晶化合物的制备方法。本发明所述的制备方法包括如下步骤:1)中间体的制备以化合物a为原料,制备中间体;2)负性液晶化合物的制备以化合物b为原料,与步骤1)所得的中间体进行反应,得到式(i)所示的负性液晶化上述制备方法中,其中,步骤2)为:以化合物b为原料,在二异丙胺、四氢呋喃和丁基锂存在的条件下,与步骤1)所得的中间体进行反应得到式(i)所示的负性液晶化合物,其反应式如下:所述的反应为在室温搅拌下反应2~4h,优选3h。所述的中间体、化合物b、二异丙胺和丁基锂的摩尔比为1:(1.0~1.2):(1.2~1.4):(1.20~1.30),优选1:1.1:1.3:1.25。所述的丁基锂和中间体在-50~-60℃下加入。具体地说,所述的步骤2)为:在反应器内加入化合物b、二异丙胺和四氢呋喃,氮气保护下,降温至-50~-60℃,滴加丁基锂,保温1~2h;控温-50~-60℃,滴加中间体,滴完升至室温搅拌2~4h得到式(i)所示的负性液晶化合物。作为优选方案,所述的搅拌后还包括后处理的过程,所述的后处理为倒入酸水水解,用乙酸乙酯提取,中和,干燥,蒸干溶剂,使用石油醚过硅胶柱,用乙醇结晶。更具体地说,所述的步骤2)为:在反应器内加入化合物b、二异丙胺和四氢呋喃,氮气保护下,降温至-50~-60℃,滴加丁基锂,保温1~2h;控温-50~-60℃,滴加中间体,滴完升至室温搅拌2~4h倒入酸水水解,用乙酸乙酯提取,中和,干燥,蒸干溶剂,使用石油醚过硅胶柱,用乙醇结晶,得到式(i)所示的负性液晶化合物。上述制备方法中,化合物b这类腈可以为已知的市售原料,大部分结构在cas已经注册,如反式丙基双环己基甲腈,cas:65355-35-3;反式-4-戊基环己基甲腈,cas:80670-47-9。也可以是采用液晶行业常用的制备方法制备得到的,如以液晶行业大量使用的烷基环己基甲酸制备酰氯,制备酰胺,然后脱水成腈。上述制备方法中,当n=1时,所述的中间体的制备按照如下步骤进行:1a)以化合物a为原料,在buli/dmf存在下,经反应得到化合物1;1b)在氟甲醚三苯基磷盐和叔丁醇钾存在下,化合物1经反应得到化合物2;1c)在盐酸存在下,化合物2经反应得到化合物3;1d)在kbh4存在下,化合物3经反应得到化合物4;1e)将化合物4与i2反应得到中间体;其合成路线如下:具体地说,步骤1a)为:四口瓶内投入化合物a、叔丁醇钾和四氢呋喃,氮气保护下,降温至-90~-100℃,滴加丁基锂溶液,然后保温,-90~-100℃下滴加n,n-二甲基甲酰胺,升至室温搅拌后将料液倒入盐酸冰水中水解,乙酸乙酯提取,干燥,过硅胶柱,得到化合物1;步骤1b)为:四口瓶内投入氯甲醚三苯基膦盐和四氢呋喃,氮气保护下,加入叔丁醇钾,然后保温,滴加化合物1和四氢呋喃配置的溶液,加完室温搅拌,反应完毕,向料液中加水终止反应,石油醚提纯,过硅胶柱,得到化合物2;步骤1c)为:四口瓶内投入化合物2,加入盐酸、水和四氢呋喃,加入回流反应,反应完毕,使用乙酸乙酯提取,然后中和,干燥,过硅胶柱,得到化合物3;步骤1d)为:四口瓶内投入化合物3、乙醇和水,分批加入硼氢化钾,加完,室温反应后将料液倒入稀盐酸水中水解,然后乙酸乙酯提取,蒸干溶剂,减压蒸馏得到化合物4;步骤1e):四口瓶内投入化合物4、三苯基膦盐、咪唑和二氯甲烷,分批加入单质碘,加完室温反应;反应完毕,加水终止反应,分液水洗,干燥蒸干二氯甲烷,用石油醚过硅胶柱,得到中间体。其中,步骤1a)中,化合物a、叔丁醇钾和丁基锂的摩尔比为1.0:1.3:1.1;步骤1b)中,氯甲醚三苯基磷盐、叔丁醇钾和化合物1的摩尔比为1.1:1.2:1.0;步骤1d)中,化合物3与硼氢化钾的摩尔比为1.0:1.0;步骤1e)中,化合物4、三苯基膦盐、咪唑和碘的摩尔比为1:1:1:1.2。当n=2时,所述的中间体的制备按照如下步骤进行:1.1)以化合物a为原料,在buli/dmf存在下,经反应得到化合物1;1.2)在氟甲醚三苯基磷盐和叔丁醇钾存在下,化合物1经反应得到化合物2;1.3)在盐酸存在下,化合物2经反应得到化合物3;1.4)化合物3经多次wittig反应、水解反应得到化合物3’;1.5)在kbh4存在下,化合物3’经反应得到化合物4’;1.6)将化合物4’与i2反应得到中间体;其合成路线如下:上述反应中,除步骤1.4)外其它与n=1时相同。上述步骤1.4)中,wittig反应为本领域常见的合成方法,用于增长碳链。采用上述技术方案后,本发明与现有技术相比具有以下有益效果:本发明所提供的负性液晶化合物侧位同时含有氰和氟,具有更强的负介电各向异性,适中的折射率,较高的清亮点,较优的化学稳定性,较优的抗uv能力。附图说明图1为本发明实施例1制得的化合物的核磁共振氢谱图;图2为本发明实施例1制得的化合物的质谱图;图3为本发明实施例18制得的化合物的核磁共振氢谱图;图4为本发明实施例18制得的化合物的质谱图;图5为本发明实施例19制得的化合物的核磁共振氢谱图;图6为本发明实施例19制得的化合物的质谱图。具体实施方式下面结合具体实施例对本发明作进一步阐述:下面实施例中gc表示气相色谱纯度,mp表示熔点,ms表示质谱;s-n:单位℃,表示液晶的晶态到向列相的熔点。△n:光学各向异性,△n=no-ne,no为寻常光的折射率,ne为非寻常光的折射率,测试条件为589nm,25±0.5℃。△ε:介电各向异性,△ε=ε∥-ε⊥,其中,ε∥为平行于分子轴的介电常数,ε⊥为垂直于分子轴的介电常数,测试条件为25±0.5℃;1khz;hp4284a;5.2微米tn左旋盒。cp:表示清亮点,该清亮点可用dsc设备直接测定而得。实施例1、制备(1)1000ml四口瓶内投入58.5g2,3-二氟苯乙醚(化合物a-1)、42g叔丁醇钾和500ml四氢呋喃。氮气保护下,降温至-90~-100℃,滴加200ml2.5mol/l的丁基锂溶液,然后保温1.5h。-90~-100℃下滴加30gn,n-二甲基甲酰胺。缓慢升至室温搅拌3h。将料液倒入盐酸冰水中水解,乙酸乙酯提取,干燥,过硅胶柱,得到4-乙氧基-2,3-二氟苯甲醛(化合物1-1),纯度97.5%(gc),产率84%。其反应式如下:(2)1000ml四口瓶内投入,氯甲醚三苯基膦盐162g,500ml四氢呋喃。氮气保护下,0℃加入叔丁醇钾58g,然后保温15min。0℃滴加80g4-乙氧基-2,3-二氟苯甲醛(化合物1-1)和100ml四氢呋喃配置的溶液。加完室温搅拌3h。反应完毕,向料液中加水终止反应,石油醚提纯,过硅胶柱,得到4-乙氧基-2,3-二氟苯乙烯醇甲醚(化合物2-1),纯度97.3%,产率85%。其反应式如下:(3)1000ml四口瓶内投入化合物2-1,加入73g30%盐酸,160ml水,160ml四氢呋喃,加入回流12h。反应完毕,使用乙酸乙酯提取,然后中和,干燥,过硅胶柱,得到4-乙氧基-2,3-二氟苯乙醛(化合物3-1),纯度84%(gc),产率70%。其反应式如下:(4)1000ml四口瓶内投入4-乙氧基-2,3-二氟苯乙醛(化合物3-1)70g,乙醇200ml,水190ml,保持0-10℃,分批加入18.4g硼氢化钾,加完,室温反应4h。将料液倒入稀盐酸水中水解,然后乙酸乙酯提取。蒸干溶解,减压蒸馏得到:4-乙氧基-2,3-二氟苯乙醇(化合物4-1)。纯度97.8%,产率64%。其反应式如下:(5)500ml四口瓶内投入29g-乙氧基-2,3-二氟苯乙醇(化合物4-1),三苯基膦盐37g,咪唑9.6g,二氯甲烷200ml。0℃分批加入单质碘11.8g,加完室温反应6h。反应完毕,加水终止反应,分液水洗,干燥蒸干二氯甲烷。有石油醚过硅胶柱,得到4-乙氧基-2,3-二氟苯乙基碘(中间体-1),纯度97.3%(gc),产率90.3%。其反应式如下:(6)500ml四口瓶内加入12.7g反式-戊基环己基甲腈(化合物b-1),二异丙胺8.3g,四氢呋喃100ml。氮气保护下,降温至-50~-60℃,滴加32ml2.5mol/l丁基锂,保温1.5h。控温-50~-60℃,滴加4-乙氧基-2,3-二氟苯乙基碘(中间体-1)20g,滴完升至室温搅拌3h。倒入酸水水解,用乙酸乙酯提取,中和,干燥,蒸干溶剂。使用石油醚过硅胶柱,用乙醇结晶得到式(i-8)所示的负性液晶化合物,纯度99.5%,产率54%。dcs:mp62.46℃。其反应式如下:对式(i-8)所示的负性液晶化合物进行结构确证,其核磁共振氢谱图如图1所示,其质谱图如图2所示。质谱图谱中分子离子峰及结构离子碎片峰显示结构符合;核磁氢谱中所含氢及对应结构氢显示结构符合。实施例2、制备(1)按照实施例1的方法制备如下式所示的中间体-1;(2)500ml四口瓶内加入化合物b-2、二异丙胺和四氢呋喃100ml。氮气保护下,降温至-50~-60℃,滴加丁基锂,保温1h。控温-50~-60℃,滴加步骤(1)所制得的中间体,其中,中间体-1、化合物b-2、二异丙胺和丁基锂的摩尔比为1:1.1:1.3:1.25,滴完升至室温搅拌2h。倒入酸水水解,用乙酸乙酯提取,中和,干燥,蒸干溶剂。使用石油醚过硅胶柱,用乙醇结晶得到上述产物。(mp:95.83-97.66℃,cp:150.75-152.0℃)实施例3、制备(1)以为原料,按照实施例1的方法制备中间体(2)以(化合物b-3)为原料,按照实施例2的方法与步骤(1)制得的中间体进行搅拌反应4h得到上述产物,其中所述的中间体、化合物b-3、二异丙胺和丁基锂的摩尔比为1:1.0:1.2:1.20。(产物mp:74.05-74.88℃cp:77.64-79.59℃)实施例4、制备(1)以为原料,按照实施例1的方法制备中间体(2)以(化合物b-4)为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物,其中所述的中间体、化合物b-4、二异丙胺和丁基锂的摩尔比为1:1.2:1.4:1.30。实施例5、制备(1)按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例6、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例7、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例8、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例9、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例10、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例11、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例12、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例13、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例14、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例15、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例16、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例17、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。实施例18、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与制得的中间体进行反应得到上述产物。对所得产物进行结构确证,其核磁共振氢谱图如图3所示,其质谱图如图4所示。质谱图谱中分子离子峰及结构离子碎片峰显示结构符合;核磁氢谱中所含氢及对应结构氢显示结构符合。实施例19、制备(1)以为原料,按照实施例1的方法制备中间体(2)以为原料,按照实施例2的方法与步骤(1)制得的中间体进行反应得到上述产物。对所得产物进行结构确证,其核磁共振氢谱图如图5所示,其质谱图如图6所示。质谱图谱中分子离子峰及结构离子碎片峰显示结构符合;核磁氢谱中所含氢及对应结构氢显示结构符合。试验例1、性能检测本试验例对本发明部分实施例所制得的负性液晶化合物的性能进行了检测,结果如下:实施例△n[589nm,20℃]△ε[khz,20℃]cp(拟合数据)实施例10.054-11.1——实施例20.086-10.14105.7℃实施例40.084-10.1137℃实施例80.058-11.44——从上述试验结果可以看出,本发明所制得的负性液晶化合物具有较好的负介电各向异性性。对本发明其它实施例制得的负性液晶化合物也进行了上述试验,其结果相似。试验例2、介电各向异性对比试验将以下单体以10%的比例添加到混合液晶wt-001中,测试不同单晶在wt-001中的介电各向异性。其中混合液晶wt-001的△ε=-4.1。如下几种单晶在同等条件下测试的△ε值如下表:混晶母液:wt-001的组成如下:液晶单体对比测试:从上述试验结果可以看出,本发明的负性液晶化合物较现有技术相比具有明显提升的负介电各向异性性能。对本发明其它实施例制得的负性液晶化合物也进行了上述试验,其结果相似。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1