具有不同官能阳离子的经表面反应的碳酸钙的后处理的制作方法
本发明涉及一种经表面反应的碳酸钙的表面处理方法,一种通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙,以及一种包含根据本发明的经表面处理的经表面反应的碳酸钙的组合物,至少一种经表面反应的碳酸钙用于在该至少一种经表面反应的碳酸钙的表面上固定至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物的用途,根据本发明的经表面处理的经表面反应的碳酸钙作为防腐剂用于控制气味和/或用于增强和/或介入基底的抗微生物活性的用途以及一种包含根据本发明的经表面处理的经表面反应的碳酸钙的制品。
背景技术:
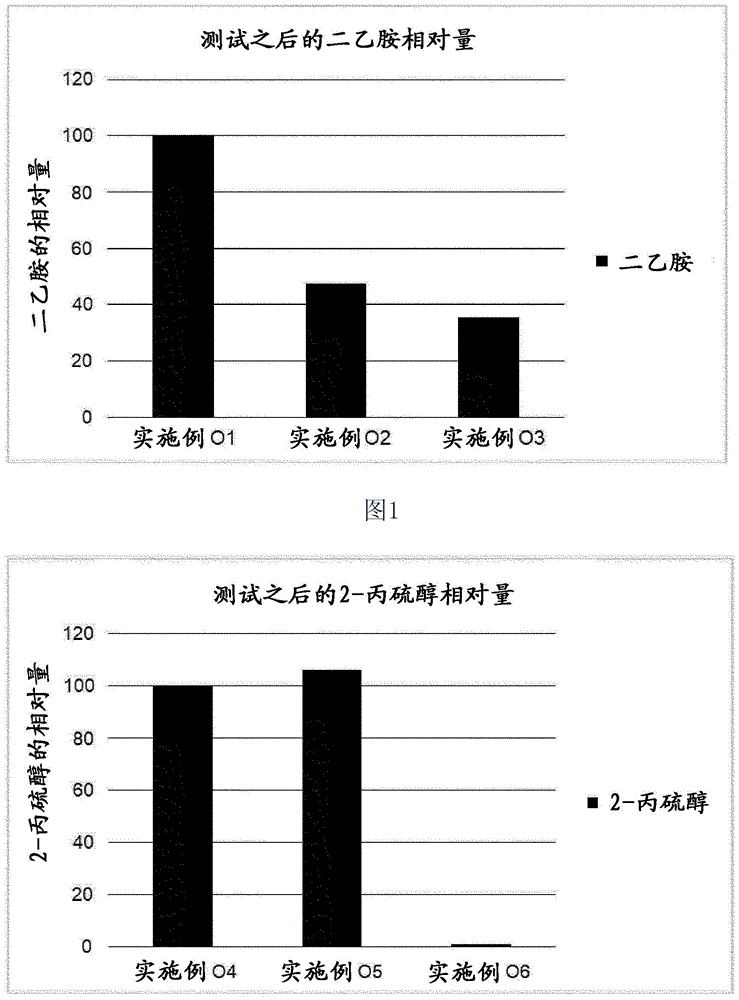
:微粒状材料上的表面处理或涂覆或吸收是化学中公知的策略,尤其是在微粒状材料的修饰和精制方面。这些策略的目标是在这些微粒状材料的表面上定位表面处理剂以改变或改善这些微粒状材料的特性,例如改善疏水性/亲水性或耐酸性。另一目标是在这些微粒的表面上定位表面处理剂以将其用作载体材料。例如,ep1084203涉及复合材料组合物,其包含至少两种矿物或有机填料或颜料以及至少一种粘结剂。该矿物或有机填料或颜料已经进行了物理或化学处理使得其具有至少一个亲有机位点。ep2029675涉及无机和/或有机微粒子以及纳米碳酸钙粒子的复合材料。借助于粘结剂涂覆这些微粒状材料的表面。kathleenlattaud等的文章“indexofrefractionenhancementofcalciteparticlescoatedwithzinccarbonate”(solidstatescience,第8卷,第10期,2006年,第1222–1228页)涉及在方解石粒子上的znco3涂层。更确切地说,水性悬浮液中的方解石粒子与氯化锌之间的化学反应促进了znco3涂层的形成,该涂层由与方解石粒子具有不同相互作用的两层构成。所获得的复合粒子具有增强的折射率。这种微粒状载体的一个应用领域为例如气味控制。一种恶臭来源例如为任何种类的废弃的人类和动物体的排泄物、液体和分泌物。然而,也存在需要被控制的其他来源的令人不愉快的气味,如那些由食品(例如来自乳制品、肉和鱼)引起的气味。关于人类或动物体液,存在控制气味的持续需求以及满足该需求的不断发展,例如在个人卫生制品如卫生巾、卫生护垫、成人失禁制品、婴儿尿布、纸巾、卫生纸和面巾纸、用于医用目的的非织造物等的领域中。这些制品常用于吸收和保留体液以及其他由人体排泄的流出物。例如,us2012/0202684涉及用于气味控制的金属离子改性的高表面积材料。该高表面积材料如纳米粒子通过在纳米粒子表面上吸收金属离子而涂覆有金属离子。所获得的悬浮液用于气味控制,例如通过使包含涂覆有金属离子的纳米粒子的悬浮液与糠基硫醇溶液混合来进行。这种微粒状载体的另一应用领域为例如作为抗微生物剂或添加剂的用途。然而,不仅微粒状材料的表面能通过便宜、简单且不耗时的方法来处理是重要的,表面处理剂在微粒状材料表面上的附着牢固或稳定也是重要的。通过在现有技术中已知的方法,表面处理剂如金属离子通常通过吸收非常弱地附着到经处理的材料的表面上,并且因此这些表面处理剂在简单的洗涤步骤中可至少部分地被洗掉。因此,经表面处理的材料可例如至少部分地失去其控制气味和/或增强抗微生物活性的能力。此外,如果表面处理剂可容易地从经表面处理的材料洗掉,则微粒状材料的改性或改善的特性如疏水性/亲水性或耐酸性会降低或失去。如果经表面处理的微粒状材料用在将会与人体接触的个人卫生制品如卫生巾、卫生护垫、成人失禁制品、婴儿尿布、纸巾、卫生纸和面巾纸、用于医用目的的非织造物等的领域中时,这是特别成问题的。通过像尿、血液或汗液的水状排泄物,表面处理剂如金属离子可从微粒状材料洗掉并且将会与皮肤接触或可经由开放性伤口进入人体。这也同样例如适用于如下这样的经表面处理的微粒状材料:该材料用于控制诸如那些由食品引起的(如来自乳制品、肉和鱼)令人不愉快的气味和/或用于赋予或增强抗微生物活性,例如在用于食品的包装产品中。如果经表面处理的微粒状材料与食品产品如肉和鱼接触,则水状挥发物如肉汁可将表面处理剂如金属离子从微粒状材料洗掉。因此,表面处理剂如金属离子可与该食品一起被摄入并且因此经口服进入人体。技术实现要素:鉴于以上所述的问题,对于提供如下这样的经表面处理的微粒状材料的方法存在持续的需求,其中表面处理剂有效地固定在微粒状材料的表面上,并且因此与通过其中表面处理剂例如仅被吸收在微粒状材料的表面上的传统方法所制备的经表面处理的微粒状材料相比,该表面处理剂更牢固地附着到微粒状材料的表面上。进一步的目的在于提供制备经表面处理的微粒状材料的方法,该方法可在成本有效、省时且生态条件下进行,即通过避免或减少有机溶剂的使用来进行。本发明的目的还在于提供可控制微生物污染但并不会对健康造成危害的材料。本发明进一步的目的在于提供除了抗微生物活性之外还具有额外益处的材料。例如,希望这种材料在延长的时间周期内赋予或增强引入其的产品的抗微生物活性,而不会以负面方式影响产品的性能。进一步的目的可从本发明的以下描述获知。前述和其他目的通过如在本文中在权利要求1中定义的主题得以解决。根据本发明的一个方面,提供经表面反应的碳酸钙的表面处理方法,包括以下步骤:(i)提供至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应,(ii)提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物,(iii)提供水性溶剂,(iv)使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液,(v)在一个或多个步骤中使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物,以及(vi)加热从步骤(v)获得的该混合物至20-250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。本发明人已经令人惊讶地发现,通过前述方法可以制备经表面处理的微粒状材料,更确切的说是具有改善的性能的经表面处理的经表面反应的碳酸钙。尤其是,通过前述方法可以制备经表面处理的经表面反应的碳酸钙,其中该表面处理剂被固定在该经表面反应的碳酸钙的孔表面以及外部上,并且因此与其中表面处理剂与经表面反应的碳酸钙的反应产物仅被吸收进微粒状材料的孔中或涂覆或吸附在微粒状材料的内或外表面上的传统微粒状材料相比,由表面处理剂(即该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物)与经表面反应的碳酸钙获得的反应产物更牢固地附着到经表面反应的碳酸钙的表面上。此外,本发明人发现,根据本发明的方法可在水中进行,并且因此在本发明的方法中可减少或避免有机溶剂。此外,根据本发明的方法可通过混合析出物(educts)来制备,并且因此可避免耗时且昂贵的中间步骤。根据本发明的另一方面,经表面处理的经表面反应的碳酸钙通过根据本发明的方法获得。根据本发明的另一方面,提供一种包含通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙的组合物。根据本发明的另一方面,至少一种经表面反应的碳酸钙用于通过根据本发明的方法制备至少一种经表面处理的经表面反应的碳酸钙在该至少一种经表面反应的碳酸钙的孔表面和外部表面上固定至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应。根据本发明又进一步的方面,提供根据本发明的经表面处理的经表面反应的碳酸钙或根据本发明的组合物作为防腐剂(preservative)用于控制气味和/或用于增强和/或介入(mediating)基底的抗微生物活性的用途。根据本发明又进一步的方面,提供根据本发明的经表面处理的经表面反应的碳酸钙或根据本发明的组合物在聚合物应用、纸涂层应用、造纸、油漆、涂料、密封剂、印刷油墨、粘合剂、食品、饲料、药物、混凝土、水泥、化妆品、水处理、工程木材应用、石膏板应用、包装应用和/或农业应用中的用途。根据本发明又进一步的方面,提供包含根据本发明的经表面处理的经表面反应的碳酸钙或根据本发明的组合物的制品,其中该制品选自纸产品、工程木材产品、石膏板产品、聚合物产品、卫生用品、医用产品、卫生保健产品、过滤产品、织造材料、非织造材料、土工织物产品、农业产品、园艺产品、衣物、鞋类产品、行李产品、家用产品、工业产品、包装产品、建筑产品、以及构造产品。本发明的有利实施方案在相应的从属权利要求中定义。根据本发明的一种实施方案,步骤(i)的该经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的15m2/g-200m2/g、优选27m2/g-180m2/g、更优选25m2/g-160m2/g且最优选30m2/g-150m2/g的比表面积,和/或(b)1-75μm、优选1.3-50μm、更优选1.5-40μm、甚至更优选1.8-30μm且最优选2-15μm的体积中值颗粒直径d50(体积),和/或(c)2-150μm、优选4-100μm、更优选6-80μm、甚至更优选8-60μm且最优选10-30μm的颗粒直径d98(体积),和/或(d)由汞孔隙率测定法测量结果计算的0.1-2.3cm3/g、更优选0.2-2.0cm3/g、尤其优选0.4-1.8cm3/g且最优选0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的另一方面,步骤(i)的该经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的30m2/g-150m2/g的比表面积,以及(b)2-15μm的体积中值颗粒直径d50(体积),以及任选地(c)10-30μm的颗粒直径d98(体积),以及任选地(d)由汞孔隙率测定法测量结果计算的0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的另一方面,步骤(i)的该经表面反应的碳酸钙以干燥形式、优选以粉末形式提供,或者以悬浮液的形式、优选以水性悬浮液的形式提供,其中该经表面反应的碳酸钙的固体含量基于该水性悬浮液的总重量计在1-80%重量、更优选3-70%重量且甚至更优选5-60%重量的范围内。根据本发明的另一方面,步骤(i)的该经表面反应的碳酸钙以进一步包含分散剂的水性悬浮液的形式提供。根据本发明的另一方面,在接触步骤(v)之前或期间将步骤(i)的该经表面反应的碳酸钙加热至20-250℃、优选25-180℃、甚至更优选50-150℃且最优选80-130℃的温度。根据本发明的另一方面,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为水溶性金属盐,优选选自铜盐、锌盐、银盐及其混合物,甚至更优选为铜盐,选自硝酸铜、硫酸铜、醋酸铜、氯化铜、氟化铜、溴化铜、其水合物及其混合物,且最优选选自硫酸铜、其水合物及其混合物。根据本发明的另一方面,步骤(iii)的该水性溶剂由水构成。根据本发明的另一方面,步骤(iv)中获得的该含金属的溶液具有基于该金属盐溶液的总重量计为1-80%重量、优选5-60%重量且最优选10-50%重量的固体含量。根据本发明的另一方面,将该至少一种金属盐、金属氢氧化物、金属氧化物或其混合物以基于该至少一种经表面反应的碳酸钙的总干重计为0.001-80%重量、优选0.01-60%重量、更优选0.05-50%重量且最优选1-40%重量的量添加到该至少一种经表面反应的碳酸钙中。根据本发明的另一方面,该接触步骤(v)通过混合和/或通过喷雾进行。根据本发明的另一方面,在步骤(vi)中将从步骤(v)获得的该混合物加热至50-180℃、优选60-160℃、甚至更优选80-150℃且最优选90-130℃的温度。根据本发明的另一方面,在步骤(v)之前和/或期间,将呈水溶液或水性悬浮液形式的碱添加到步骤(i)的该至少一种经表面反应的碳酸钙中,其中该碱优选包含碳酸根离子且最优选该碱为碳酸钠。根据本发明的另一方面,该碱以基于该至少一种经表面反应的碳酸钙的总干重计为0.001-80%重量、优选0.01-60%重量、更优选0.05-50%重量且最优选1-40%重量的量添加。根据本发明的另一方面,步骤(vi)中获得的该经表面处理的经表面反应的碳酸钙为水性悬浮液的形式,该方法进一步包括步骤(vii):在步骤(iv)之后从水性悬浮液分离从步骤(vi)获得的该经表面处理的经表面反应的碳酸钙。根据本发明的另一方面,该方法进一步包括在步骤(vi)之前、期间或之后研磨和/或分级和/或分类从步骤(v)获得的该混合物的步骤。根据本发明的另一方面,该方法进一步包括如下的步骤:其中在步骤(vi)中形成的该经表面处理的经表面反应的碳酸钙进行后处理,优选在如果存在的步骤(vii)、(viii)、(ix)或(x)之后,并且优选利用脂肪酸如硬脂酸、硅烷或脂肪酸的磷酸酯或硅氧烷进行后处理。根据本发明的另一方面,该方法进一步包括步骤(viii):洗涤从步骤(vi)或如果存在的步骤(vii)获得的该经表面处理的经表面反应的碳酸钙。根据本发明的另一方面,该方法进一步包括步骤(ix):在步骤(vi)或如果存在的步骤(vii)或(viii)之后,在60-600℃、优选80-550℃的温度下干燥该经表面处理的经表面反应的碳酸钙。根据本发明的另一方面,该方法进一步包括步骤(x):在加热步骤(vi)之前机械脱水从步骤(v)获得的该混合物,优选通过离心或过滤进行。根据本发明的另一方面,该方法为间歇或连续方法,优选连续方法。根据本发明的另一方面,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为硫酸铜、其水合物或其混合物和/或在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物为水合磷酸氢铜(cuhpo4·h2o)。根据本发明的另一方面,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为硫酸锌和/或氯化锌、其水合物或其混合物。根据本发明的另一种实施方案,该经表面处理的经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的15m2/g-200m2/g、优选27m2/g-180m2/g、更优选25m2/g-160m2/g且最优选30m2/g-150m2/g的比表面积,和/或(b)1-75μm、优选1.3-50μm、更优选1.5-40μm、甚至更优选1.8-30μm且最优选2-15μm的体积中值颗粒直径d50(体积),和/或(c)2-150μm、优选4-100μm、更优选6-80μm、甚至更优选8-60μm且最优选10-30μm的颗粒直径d98(体积),和/或(d)由汞孔隙率测定法测量结果计算的0.1-2.3cm3/g、更优选0.2-2.0cm3/g、尤其优选0.4-1.8cm3/g且最优选0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的另一种实施方案,该经表面处理的经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的30m2/g-150m2/g的比表面积,以及(b)2-15μm的体积中值颗粒直径d50(体积),以及任选地(c)10-30μm的颗粒直径d98(体积),以及任选地(d)由汞孔隙率测定法测量结果计算的0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的另一种实施方案,该经表面处理的经表面反应的碳酸钙包含至少一种选自水合磷酸氢铜(cuhpo4·h2o),孔雀石(cu2co3(oh)2),水胆矾(cu4so4(oh)6),钙铜矾(cacu4(so4)2(oh)6·3h2o),一水蓝铜矾(cu4(so4)(oh)6·h2o)及其混合物的化合物作为在该经表面反应的碳酸钙的表面上形成的水不溶性金属化合物。根据本发明的另一种实施方案,该经表面处理的经表面反应的碳酸钙包含在该经表面反应的碳酸钙的表面上的至少一种不溶性锌盐。根据本发明的另一种实施方案,有气味的物质(odorant)选自胺如三乙胺、二乙胺、三甲胺、二氨基丁烷、四亚甲基二胺、五亚甲基二胺、吡啶、吲哚、3-甲基吲哚;羧酸如丙酸、丁酸、3-甲基丁酸、2-甲基丙酸、己酸;硫有机化合物如硫醇如甲硫醇,磷有机化合物如甲膦、二甲膦、其衍生物及其混合物;优选该有气味的物质为胺且最优选该有气味的物质为二乙胺。应理解,出于本发明的目的,以下术语具有以下含义:根据本发明的术语“表面处理剂”是用于处理该至少一种经表面反应的碳酸钙的表面的试剂。本发明的表面处理剂为水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。在本发明主旨中的“经表面处理的经表面反应的碳酸钙”包含至少一种经表面反应的碳酸钙,其已经与根据本发明的至少一种表面处理剂接触以便获得位于该经表面反应的碳酸钙的表面的至少一部分上的处理层。因此,术语“处理层”是指在该经表面反应的碳酸钙的表面的至少一部分上的包含该表面处理剂及其反应产物的层。在本发明含义中的术语“反应产物”是指通过使该至少一种经表面反应的碳酸钙与根据本发明的至少一种表面处理剂接触获得的产物。更确切的说,术语“反应产物”至少包括在该经表面反应的碳酸钙的表面上由作为金属盐、金属氢氧化物、金属氧化物及其混合物提供的金属形成的水不溶性金属化合物。该表面处理剂与该经表面反应的碳酸钙的反应产物以湿以及干燥状态彼此化学或物理地结合,这意味着没有观察到离析(segregation)。根据本发明的术语“碱”是指如由布朗斯台德和洛瑞理论所定义的碱。因此,在本发明含义中的碱是能接受氢离子(h+)(也称为质子)的物质。在本发明含义中的“含碳酸钙材料”是矿物材料或合成材料,其具有基于该含碳酸钙材料的总重量计为至少50%重量、优选至少75%重量、更优选至少90%重量且最优选至少95%重量的碳酸钙含量。在本发明含义中的“研磨碳酸钙”(gcc)为自天然来源(例如石灰石、大理石或白垩)获得的碳酸钙,并且其通过湿式和/或干式处理如研磨、筛选和/或分级(例如借助于旋风器或分级器)进行加工。在本发明含义中的“沉淀碳酸钙”(pcc)为合成材料,通常通过在水性环境中在二氧化碳与氢氧化钙(熟石灰)反应之后沉淀或通过钙和碳酸根源在水中沉淀而获得。另外可选地,沉淀碳酸钙还可以是在水性环境中引入钙和碳酸盐如氯化钙和碳酸钠的产物。pcc可具有球霰石、方解石或文石晶型。pcc被描述于例如以下文献中:ep2447213a1、ep2524898a1、ep2371766a1、ep2840065a1或wo2013/142473a1。根据本发明的“经表面反应的碳酸钙”是天然研磨碳酸钙或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体的反应产物,其中该二氧化碳通过h3o+离子供体原位形成和/或从外部来源供应。在本发明的上下文中,h3o+离子供体是布朗斯台德酸和/或酸式盐。术语“干(燥)(dry)”或“(经)干燥的(dried)”材料被理解为是指具有基于该材料重量的总重量计(例如基于该经表面处理的经表面反应的碳酸钙重量的总重量计)为0.001-30%重量的水的材料。水%(等于“含湿量(moisturecontent)”)根据coulometrickarlfischer测量方法确定,其中将填料材料加热至220℃,并且作为水蒸气释放且使用氮气流(在100ml/min下)分离的水含量在coulometrickarlfischer单元中确定。在本发明意义上的“干燥(drying)”是指进行加热,直至该材料(例如该经表面处理的经表面反应的碳酸钙)的含湿量基于该经表面处理的经表面反应的碳酸钙重量的总重量计为0.001-30%重量为止。除本文的经表面反应的碳酸钙或经表面处理的经表面反应的碳酸钙之外的微粒状材料的“粒子尺寸”通过其粒子尺寸的分布dx来描述。在其中,值dx表示下述这样的直径:相对于该直径,x%重量的粒子具有小于dx的直径。这意味着例如d20值是指下述这样的粒子尺寸:其中所有粒子的20%重量小于该粒子尺寸。d50值因而是重量中值粒子尺寸,也即所有颗粒的50%重量大于该粒子尺寸且剩余的50%重量小于该粒子尺寸。出于本发明的目的,除非另外指明,否则该粒子尺寸被具体指定为重量中值粒子尺寸d50。d98值是指下述这样的粒子尺寸:其中所有粒子的98%重量小于该粒子尺寸。粒子尺寸通过使用micromeriticsinstrumentcorporation的sedigraphtm5100或5120仪器来确定。方法及仪器为本领域技术人员所知且通常用于确定填料和颜料的粒子尺寸。在0.1%重量的na4p2o7的水溶液中进行测量。使用高速搅拌机和超声分散样品。使用malvernmastersizer2000激光衍射系统评价体积中值颗粒直径d50。使用malvernmastersizer2000激光衍射系统测量的d50或者d98值所表示的直径值使得以体积计分别为50%或者98%的粒子具有小于该值的直径。使用米氏(mie)理论分析通过测量结果获得的原始数据,其中粒子折射率为1.57并且吸收系数为0.005。在本发明含义中的经表面反应的碳酸钙以及经表面处理的经表面反应的碳酸钙的“比表面积(ssa)”被定义为该碳酸钙的表面积除以其质量。如在本文中所用的比表面积通过氮气吸附使用bet等温线(iso9277:2010)进行测量并且具体指定为m2/g。在本发明含义中的术语“表面积”或“外表面”是指如用于根据iso9277:2010测量bet的氮气可及的经表面反应的碳酸钙粒子的表面。在这方面,应当注意,该表面积完全饱和所需的根据权利要求1的表面处理剂的量被定义为单层浓度。因而可通过在该经表面反应的碳酸钙粒子的表面上形成双层或多层结构来选择更高浓度。出于本发明的目的,粒子内比孔容被表示为由压力下的汞侵入比在每单位重量样品的离散双峰孔尺寸分布中的拐点更小的孔的体积。相应的孔隙率进而被表示为经侵入的体积/单位体积的样品。侵入式孔隙率测定法测量结果采用micromeriticsautoporev9620汞孔率计,该汞孔率计具有414mpa(60000psi)的最大施加汞压,等效于0.004μm的拉普拉斯喉径。出于本发明的目的,“水不溶性”材料被定义为这样的材料:当100g所述材料与100g去离子水混合且在20℃下在具有0.2μm孔尺寸的过滤器上过滤以回收液体滤液时,在环境压力下100g所述液体滤液在95-100℃下蒸发之后提供小于或等于1g的回收的固体材料。“水溶性”材料被定义为这样的材料:当100g所述材料与100g去离子水混合且在20℃下在具有0.2μm孔尺寸的过滤器上过滤以回收液体滤液时,在环境压力下100g所述液体滤液在95-100℃下蒸发之后提供大于1g的回收的固体材料。在本发明含义中的“悬浮液”或“浆料”包含不溶性固体和溶剂或液体(优选水)以及任选的其他添加剂,且通常包含大量的固体,并且因而与形成其的液体相比更为粘稠且可具有更高的密度。根据本发明的术语“固体”是指在标准环境温度和压力(satp)下为固体的材料,该标准环境温度和压力(satp)是指298.15k(25℃)的温度和精确地为100000pa(1巴,14.5psi,0.98692atm)的绝对压力。该固体可呈粉末、片剂、粒料、薄片等形式。根据本发明的“有气味的物质(odorant)”是具有嗅味(smell)或气味(odour)的化合物,即挥发性足以输送至鼻子上部的嗅觉系统中。出于本发明的目的,术语“粘度”或“布氏粘度”是指布氏粘度。该布氏粘度为此目的通过布氏dv-iiiultra粘度计在24℃±3℃下在100rpm下使用布氏rv-锭子组的合适锭子来测量,且具体指定为mpa·s。一旦锭子已经被插入到样品中,则以100rpm的恒定旋转速度开始测量。所报告的布氏粘度值是在测量开始之后60秒显示的值。本领域技术人员基于其技术知识将从布氏rv-锭子组中选择适合于待测量的粘度范围的锭子。例如,对于200-800mpa·s之间的粘度范围,可使用锭子编号3,对于在400-1600mpa·s之间的粘度范围,可使用锭子编号4,对于800-3200mpa·s之间的粘度范围,可使用锭子编号5,对于1000-2000000mpa·s之间的粘度范围,可使用锭子编号6,且对于4000-8000000mpa·s之间的粘度范围,可使用锭子编号7。当术语“包括或包含(comprising)”在本说明书和权利要求书中被使用时,其并不排除其他未具体指出的具有主要或次要功能重要性的要素。出于本发明的目的,术语“由……构成(consistingof)”被认为是术语“包括或包含(comprisingof)”的优选实施方案。如果在下文中定义一个组集(group)包括至少一定数目的实施方案,则这也被理解为公开了一个组集,其优选仅由这些实施方案构成。无论何处使用术语“包括或包含(including)”或者“具有(having)”,这些术语被认为等同于如上定义的“包括或包含(comprising)”。在谈论单数名词时使用不定冠词或定冠词如“a”、“an”或“the”的情况下,这包括了该名词的复数,除非一些情况下另外具体指出。诸如“可获得(obtainable)”或“可定义(definable)”以及“获得(的)(obtained)”或“定义(的)(defined)”的术语可互换使用。这例如意味着,除非上下文另外明确指出,否则术语“获得(的)”并不意味着指示例如一种实施方案必须通过例如术语“获得(的)”之后的步骤序列来获得,虽然术语“获得(的)”或“定义(的)”总是包括此类限制性理解作为优选实施方案。附图说明图1涉及在有气味的物质测试之后二乙胺的相对量。图2涉及在有气味的物质测试之后2-丙硫醇的相对量。具体实施方式如以上所阐述的,用于表面处理经表面反应的碳酸钙的本发明方法至少包括方法步骤(i)、(ii)、(iii)、(iv)、(v)和(vi)。下文涉及本发明并且尤其是用于表面处理经表面反应的碳酸钙的本发明方法的前述步骤的进一步细节。本发明的方法可以连续方法或间歇方法的形式进行,优选以连续方法的形式进行。步骤(i)的表征:提供经表面反应的碳酸钙根据本发明方法的步骤(i),提供经表面反应的碳酸钙。应理解,该经表面反应的碳酸钙可为一种或多种经表面反应的碳酸钙。在本发明的一种实施方案中,该经表面反应的碳酸钙包括一种类型的经表面反应的碳酸钙,优选由一种类型的经表面反应的碳酸钙构成。另外可选地,该经表面反应的碳酸钙包括两种或更多种类型的经表面反应的碳酸钙,优选由两种或更多种类型的经表面反应的碳酸钙构成。例如,该经表面反应的碳酸钙包括两种或三种类型的经表面反应的碳酸钙,优选由两种或三种类型的经表面反应的碳酸钙构成。优选地,该经表面反应的碳酸钙包括一种类型的经表面反应的碳酸钙,更优选由一种类型的经表面反应的碳酸钙构成。该经表面反应的碳酸钙是天然研磨碳酸钙或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应。在本发明上下文中的h3o+离子供体为布朗斯台德酸和/或酸式盐。在本发明的优选实施方案中,该经表面反应的碳酸钙通过包括以下步骤的方法获得:(a)提供天然或沉淀碳酸钙的悬浮液,(b)向步骤a)的该悬浮液中添加至少一种在20℃下具有0或更小的pka值或者在20℃下具有0-2.5的pka值的酸,以及(c)在步骤(b)之前、期间或之后用二氧化碳处理步骤(a)的该悬浮液。根据另一实施方案,该经表面反应的碳酸钙通过包括以下步骤的方法获得:(a)提供天然或沉淀碳酸钙,(b)提供至少一种水溶性酸,(c)提供气态co2,(d)使步骤(a)的所述天然或沉淀碳酸钙与步骤(b)的该至少一种酸以及步骤(c)的co2接触,其特征在于:(i)步骤b)的该至少一种酸在20℃下具有大于2.5且小于或等于7的pka,该pka与其第一可用氢的电离相关,且在此第一可用氢失去时形成能够形成水溶性钙盐的相应阴离子,且(ii)在使该至少一种酸与天然或沉淀碳酸钙接触之后,额外提供至少一种水溶性盐,其在含氢盐情形下在20℃下具有大于7的pka(该pka与该第一可用氢的电离相关),且其盐阴离子能够形成水不溶性钙盐。“天然研磨碳酸钙(gcc)”优选选自含碳酸钙的矿物,所述含碳酸钙的矿物选自大理石、白垩、白云石、石灰石及其混合物。天然研磨碳酸钙可进一步包含天然存在的组分例如碳酸镁、铝硅酸盐等。通常,天然研磨碳酸钙的研磨可以是干或湿研磨步骤并且可例如在使得粉碎主要由使用辅助体冲击产生的条件下,用任何传统研磨装置进行,也即在以下的一种或多种中进行:球磨机、棒磨机、振动研磨机、轧碎机、离心冲击研磨机、立式珠磨机、磨碎机、销棒粉碎机、锤磨机、粉磨机、撕碎机、去块机、切割机(knifecutter)或本领域技术人员已知的其他此类设备。在含碳酸钙的矿物材料包含湿研磨的含碳酸钙的矿物材料的情况下,研磨步骤可在使得发生自体研磨的条件下和/或通过水平球磨和/或本领域技术人员已知的其他此类方法来进行。由此获得的经湿加工的研磨的含碳酸钙的矿物材料可通过众所周知的方法,例如通过絮凝、过滤或强制蒸发(在干燥之前)来洗涤并脱水。后续干燥步骤(必要时)可在单一步骤(例如喷雾干燥)中进行,或者在至少两个步骤中进行。还常见地,这种矿物材料进行选矿步骤(例如浮选、漂白或磁性分离步骤)以去除杂质。在本发明含义中的“沉淀碳酸钙”(pcc)为合成材料,通常通过在水性环境中在二氧化碳与氢氧化钙反应之后沉淀或通过将钙离子和碳酸根离子(例如,cacl2及na2co3)从溶液中沉淀出来而获得。生产pcc的其他可能方式为石灰纯碱法,或者solvay法,其中pcc为氨生产的副产物。沉淀碳酸钙以三种初级晶形存在:方解石、文石和球霰石,且对于这些晶形中的每一晶形而言存在许多不同的多晶型物(晶体惯态)。方解石具有三角结构,该三角结构具有典型的晶体惯态如偏三角面体的(s-pcc)、斜方六面体的(r-pcc)、六角形棱柱的、轴面的、胶体的(c-pcc)、立方的及棱柱的(p-pcc)。文石为正斜方晶结构,该正斜方晶结构具有成对六角形棱晶的典型晶体惯态,以及细长棱柱的、弯曲叶片状的、陡锥状、凿尖晶体、分叉树及珊瑚或蠕虫状的形式的多种分类。球霰石属于六方晶系。所获得的pcc浆料可机械脱水和干燥。根据本发明的一种实施方案,该沉淀碳酸钙为优选包含文石、球霰石或方解石矿物晶形或其混合物的沉淀碳酸钙。沉淀碳酸钙可以在用二氧化碳和至少一种h3o+离子供体处理之前通过与用于研磨如上所述的天然碳酸钙的相同方式进行研磨。根据本发明的一种实施方案,天然研磨或沉淀碳酸钙为粒子的形式,所述粒子具有0.05-10.0μm、优选0.2-5.0μm、更优选0.4-3.0μm、最优选0.6-1.2μm、尤其是0.7μm的重量中值粒子尺寸d50。根据本发明的另一种实施方案,天然或沉淀碳酸钙为粒子的形式,所述粒子具有0.15-55μm、优选1-40μm、更优选2-25μm、最优选3-15μm、尤其是4μm的顶切粒子尺寸d98。该天然和/或沉淀碳酸钙可干燥使用或悬浮在水中。优选地,相应的浆料具有基于该浆料的重量计在1%重量-90%重量、更优选3%重量-60%重量、甚至更优选5%重量-40%重量并且最优选10%重量-25%重量范围内的天然或沉淀碳酸钙的含量。用于制备经表面反应的碳酸钙的该一种或多种h3o+离子供体可为在制备条件下产生h3o+离子的任何强酸、中强酸或弱酸或其混合物。根据本发明,该至少一种h3o+离子供体也可以是在制备条件下产生h3o+离子的酸式盐。根据一种实施方案,该至少一种h3o+离子供体为具有在20℃下为0或更小的pka的强酸。根据另一种实施方案,该至少一种h3o+离子供体为在20℃下pka值为0-2.5的中强酸。如果在20℃下的pka为0或更小,则该酸优选选自硫酸,盐酸或者其混合物。如果在20℃下的pka为0-2.5,则h3o+离子供体优选选自h2so3、h3po4、草酸或其混合物。该至少一种h3o+离子供体也可以是酸式盐,例如hso4-或h2po4-(其为通过相应阳离子如li+、na+或k+至少部分中和的),或者hpo42-(其为通过相应阳离子如li+、na+、k+、mg2+或ca2+至少部分中和的)。该至少一种h3o+离子供体也可以是一种或多种酸和一种或多种酸式盐的混合物。根据又另一种实施方案,该至少一种h3o+离子供体为弱酸,其具有在20℃下测量时为大于2.5且小于或等于7的pka值(与第一可用氢的电离相关),并且具有能够形成水溶性钙盐的相应阴离子。之后,额外提供至少一种水溶性盐,其在含氢盐的情况下具有在20℃下测量时为大于7的pka(与第一可用氢的电离相关)并且其盐阴离子能够形成水不溶性钙盐。根据该优选的实施方案,该弱酸具有在20℃下大于2.5至5的pka值,并且更优选地,该弱酸选自醋酸、甲酸、丙酸及其混合物。所述水溶性盐的示例性阳离子选自钾、钠、锂及其混合物。在一种更优选的实施方案中,该阳离子为钠或钾。所述水溶性盐的示例性阴离子选自磷酸根、磷酸二氢根、磷酸单氢根、草酸根、硅酸根、其混合物及其水合物。在一种更优选的实施方案中,所述阴离子选自磷酸根、磷酸二氢根、磷酸单氢根、其混合物及其水合物。在一种最优选的实施方案中,所述阴离子选自磷酸二氢根、磷酸单氢根、其混合物及其水合物。可逐滴或在一个步骤中进行水溶性盐的添加。在逐滴添加的情况下,该添加优选在10分钟的时间周期内发生。更优选地,在一个步骤中添加所述盐。根据本发明的一种实施方案,该至少一种h3o+离子供体选自盐酸、硫酸、亚硫酸、磷酸、柠檬酸、草酸、醋酸、甲酸及其混合物。优选地,该至少一种h3o+离子供体选自盐酸、硫酸、亚硫酸、磷酸、草酸、h2po4-(其为通过相应阳离子如li+、na+或k+至少部分中和的),hpo42-(其为通过相应阳离子如li+、na+、k+、mg2+或ca2+至少部分中和的),及其混合物,更优选地,该至少一种酸选自盐酸、硫酸、亚硫酸、磷酸、草酸或其混合物,且最优选地,该至少一种h3o+离子供体为磷酸。该一种或多种h3o+离子供体可作为浓缩溶液或更稀释的溶液被添加至该悬浮液中。优选地,该h3o+离子供体与该天然或沉淀碳酸钙的摩尔比为0.01-4,更优选0.02-2,甚至更优选0.05-1并且最优选0.1-0.58。作为另外可选方案,也可在悬浮该天然或沉淀碳酸钙之前将该h3o+离子供体添加至水中。在下一个步骤中,利用二氧化碳处理天然或沉淀碳酸钙。如果诸如硫酸或盐酸的强酸被用于天然或沉淀碳酸钙的h3o+离子供体处理,则二氧化碳自动形成。另外可选地或者额外地,二氧化碳可从外部来源供应。h3o+离子供体处理和利用二氧化碳的处理可同时进行,当使用强酸或中强酸时情况就是如此。也可首先进行h3o+离子供体处理,例如利用在20℃下具有0-2.5范围内的pka的中强酸来进行,其中二氧化碳原位形成,并且因此,二氧化碳处理将自动地与h3o+离子供体处理同时进行,接着进行利用从外部来源供应的二氧化碳的额外处理。优选地,悬浮液中气态二氧化碳的浓度就体积而言使得比率(悬浮液体积):(气态co2的体积)为1:0.05至1:20,甚至更优选为1:0.05至1:5。在一种优选实施方案中,h3o+离子供体处理步骤和/或二氧化碳处理步骤重复至少一次,更优选多次。根据一种实施方案,在至少约5分钟、优选至少约10分钟、典型地约10-约20分钟、更优选约30分钟、甚至更优选约45分钟并且有时约1小时或更长的时间周期内添加该至少一种h3o+离子供体。在h3o+离子供体处理及二氧化碳处理之后,在20℃下测量的水性悬浮液的ph值自然地达到大于6.0、优选大于6.5、更优选大于7.0、甚至更优选大于7.5的值,由此将表面改性的天然或沉淀碳酸钙制备为具有大于6.0、优选大于6.5、更优选大于7.0、甚至更优选大于7.5的ph的水性悬浮液。关于经表面反应的天然碳酸钙的制备的进一步细节公开在以下文献中:wo00/39222a1,wo2004/083316a1,wo2005/121257a2,wo2009/074492a1,ep2264108a1,ep2264109a1和us2004/0020410a1,这些参考文献的内容在此被包括在本申请中。类似地,获得经表面反应的沉淀碳酸钙。正如可从wo2009/074492a1详细获取的,经表面反应的沉淀碳酸钙通过如下方式获得:使沉淀碳酸钙与h3o+离子并且与溶解于水性介质中且能够形成水不溶性钙盐的阴离子在水性介质中接触,以形成经表面反应的沉淀碳酸钙的浆料,其中所述经表面反应的沉淀碳酸钙包含在沉淀碳酸钙的至少一部分的表面上形成的所述阴离子的不溶性的、至少部分结晶的钙盐。所述溶解的钙离子对应于相对于沉淀碳酸钙被h3o+离子溶解而自然生成的溶解钙离子来说过量的溶解的钙离子,其中所述h3o+离子仅以该阴离子的抗衡离子的形式提供,也即经由添加酸或非钙酸式盐的形式的阴离子,并且不存在任何另外的钙离子或钙离子生成来源。所述过量的溶解的钙离子优选通过添加可溶中性或酸式钙盐而提供,或通过添加原位产生可溶中性或酸式钙盐的酸或中性或酸式非钙盐而提供。所述h3o+离子可通过添加酸或所述阴离子的酸式盐而提供,或通过添加同时用于提供所有或部分的所述过量的溶解的钙离子的酸或酸式盐而提供。在制备经表面反应的天然或沉淀碳酸钙的一种进一步优选的实施方案中,使天然或沉淀碳酸钙在至少一种选自以下的化合物的存在下与一种或多种h3o+离子供体和/或二氧化碳反应:硅酸盐、二氧化硅、氢氧化铝、碱土铝酸盐如铝酸钠或铝酸钾、氧化镁或其混合物。优选地,该至少一种硅酸盐选自硅酸铝、硅酸钙或碱土金属硅酸盐。这些组分可在添加一种或多种h3o+离子供体和/或二氧化碳之前添加至包含天然或沉淀碳酸钙的水性悬浮液中。另外可选地,硅酸盐和/或二氧化硅和/或氢氧化铝和/或碱土铝酸盐和/或氧化镁组分可在天然或沉淀碳酸钙与一种或多种h3o+离子供体及二氧化碳已经开始反应时添加至天然或沉淀碳酸钙的水性悬浮液中。关于在至少一种硅酸盐和/或二氧化硅和/或氢氧化铝和/或碱土铝酸盐组分存在下制备经表面改性的天然或沉淀碳酸钙的其他细节被公开于wo2004/083316a1中,该参考文献的内容在此被包含在本申请中。可使经表面反应的碳酸钙保持悬浮液状态,任选地通过分散剂使其进一步稳定化。可使用本领域技术人员已知的传统分散剂。优选的分散剂包括聚丙烯酸和/或羧甲基纤维素。另外可选地,可干燥上述水性悬浮液,由此获得呈粒料或粉末形式的固体(也即干燥的或含有不呈流体形式的很少的水)经表面反应的天然或沉淀碳酸钙。经表面反应的碳酸钙可具有不同的粒子形状,例如玫瑰、高尔夫球和/或大脑的形状。在一种优选的实施方案中,该经表面反应的碳酸钙具有使用氮气和bet方法测量的15m2/g-200m2/g、优选27m2/g-180m2/g、更优选25m2/g-160m2/g且最优选30m2/g-150m2/g的比表面积。例如,该经表面反应的碳酸钙具有使用氮气和bet方法测量的30m2/g-100m2/g的比表面积。在本发明的含义中,bet比表面积被定义为粒子的表面积除以粒子的质量。如在本文中使用的,比表面积使用bet等温线通过吸附来测量(iso9277:2010)且具体指定为m2/g。此外优选地,该经表面反应的碳酸钙粒子具有1-75μm、优选1.3-50μm、更优选1.5-40μm、甚至更优选1.8-30μm且最优选2-15μm的体积中值颗粒直径d50(体积)。此外可优选该经表面反应的碳酸钙粒子具有2-150μm、优选4-100μm、更优选6-80μm、甚至更优选8-60μm且最优选10-30μm的颗粒直径d98(体积)。值dx表示下述这样的直径:相对于该直径,x%的粒子具有小于dx的直径。这意味着d98值是指下述这样的粒子尺寸:其中所有粒子的98%小于该粒子尺寸。d98值也被称作“顶切(topcut)”。dx值可以用体积或重量百分数给出。d50(重量)值因而是重量中值粒子尺寸,也即所有颗粒的50%重量小于该粒子尺寸,并且d50(体积)值是体积中值粒子尺寸,也即所有颗粒的50%体积小于该粒子尺寸。使用malvernmastersizer2000激光衍射系统评价体积中值颗粒尺寸d50。使用malvernmastersizer2000激光衍射系统测量的d50或d98值指示出的直径值为:分别50%或98%体积的粒子具有小于这个值的直径。通过测量获得的原始数据使用米氏理论、以粒子折射率1.57以及吸收系数0.005进行分析。重量中值颗粒尺寸通过沉降法测量,所述沉降法是在重力场中的沉降行为的分析。使用micromeriticsinstrumentcorporation的sedigraphtm5100或5120进行测量。方法及仪器为本领域技术人员所知且通常用于确定填料和颜料的颗粒尺寸。在0.1%重量na4p2o7的水溶液中进行测量。使用高速搅拌器及超声分散样品。方法及仪器为本领域技术人员所知且通常用于确定填料和颜料的颗粒尺寸。使用micromeriticsautoporev9620汞孔率计,使用汞侵入孔隙率测定法(mercuryintrusionporosimetry)测量结果测量比孔容,所述汞孔率计具有最大施加汞压为414mpa(60000psi),等效于0.004μm(~nm)的拉普拉斯喉径。在每个压力步骤使用的平衡时间是20秒。将样品材料密封在3cm3室的粉末透度计中用于分析。使用软件pore-comp(gane,p.a.c.,kettle,j.p.,matthews,g.p.和ridgway,c.j.,“voidspacestructureofcompressiblepolymerspheresandconsolidatedcalciumcarbonatepaper-coatingformulations”,industrialandengineeringchemistryresearch,35(5),1996年,第1753-1764页),针对汞压缩、透度计膨胀和样品材料压缩来校正数据。在累积侵入数据中见到的总孔体积可被分成两个区域,其中从214μm降至约1-4μm的侵入数据显示具有强烈贡献的任何附聚结构之间的样品的粗填充。在这些直径之下的是粒子自身的精细粒子间填充。如果它们也具有粒子内孔,则此区域显现双峰,并且通过获取由汞侵入比峰转折点更细(即比双峰拐点更细)的孔的比孔容,因而定义比粒子内孔体积。这三个区域的总和给出了粉末的总全部孔体积,但强烈地取决于原始样品压实/在分布的粗孔末端处的粉末的沉降。通过获取累积侵入曲线的第一导数,揭示了基于等效拉普拉斯直径的孔尺寸分布,其必然包括孔屏蔽。微分曲线清楚地显示了粗附聚孔结构区域、粒子间孔区域和粒子内孔区域(如果存在的话)。已知粒子内孔直径范围,则可以从总孔体积中减去剩余粒子间和附聚体间孔体积,以给出在每单位质量孔体积(比孔容)方面的单独的内部孔的希望的孔体积。当然,相同的减法原理也适用于分离任何感兴趣的其他孔尺寸区域。优选地,该经表面反应的碳酸钙具有由汞孔隙率测定法测量结果计算的0.1-2.3cm3/g、更优选0.2-2.0cm3/g、尤其优选0.4-1.8cm3/g且最优选0.6-1.6cm3/g的粒子内侵入式比孔容。通过汞孔隙率测定法测量结果确定的该经表面反应的碳酸钙的粒子内孔尺寸优选0.004-1.6μm、更优选0.005-1.3μm、尤其优选0.006-1.15μm且最优选0.007-1.0μm,例如0.01-0.9μm。根据本发明的一种实施方案,步骤(i)的该经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的15m2/g-200m2/g、优选27m2/g-180m2/g、更优选25m2/g-160m2/g且最优选30m2/g-150m2/g的比表面积,和/或(b)1-75μm、优选1.3-50μm、更优选1.5-40μm、甚至更优选1.8-30μm且最优选2-15μm的体积中值颗粒直径d50(体积),和/或(c)2-150μm、优选4-100μm、更优选6-80μm、甚至更优选8-60μm且最优选10-30μm的颗粒直径d98(体积),和/或(d)由汞孔隙率测定法测量结果计算的0.1-2.3cm3/g、更优选0.2-2.0cm3/g、尤其优选0.4-1.8cm3/g且最优选0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的另一种实施方案,步骤(i)的该经表面反应的碳酸钙具有(a)根据iso9277使用氮气和bet方法测量的30m2/g-150m2/g的比表面积,以及(b)2-15μm的体积中值颗粒直径d50(体积),以及任选地(c)10-30μm的颗粒直径d98(体积),以及任选地(d)由汞孔隙率测定法测量结果计算的0.6-1.6cm3/g的粒子内侵入式比孔容。应理解,该经表面反应的碳酸钙可以水性悬浮液的形式或以干燥形式提供。如果该经表面反应的碳酸钙以水性悬浮液的形式提供,则该水性悬浮液优选具有基于该水性悬浮液的总重量计为1-80%重量的固体含量。根据一种优选的实施方案,该水性悬浮液的固体含量基于该水性悬浮液的总重量计为3-70%重量,并且甚至更优选为5-60%重量。术语“水性”悬浮液是指一种体系,其中液相包含水,优选由水构成。然而,所述术语并不排除该水性悬浮液的液相包含少量的至少一种选自甲醇、乙醇、丙酮、乙腈、四氢呋喃及其混合物的水混溶性有机溶剂。如果该水性悬浮液包含至少一种水混溶性有机溶剂,则该水性悬浮液的液相以基于该水性悬浮液的液相的总重量计为0.1-40.0%重量、优选0.1-30.0%重量、更优选0.1-20.0%重量且最优选0.1-10.0%重量的量包含该至少一种水混溶性有机溶剂。例如,该水性悬浮液的液相由水构成。根据一种优选的实施方案,该水性悬浮液由水和该经表面反应的碳酸钙构成。另外可选地,该水性的经表面反应的碳酸钙的悬浮液包含其他的添加剂。额外地或另外可选地,该水性的经表面反应的碳酸钙的悬浮液包含分散剂如聚丙烯酸酯。优选地,步骤a)中提供的该经表面反应的碳酸钙为干燥的经表面反应的碳酸钙。这种实施方案是有利的,因为该方法可在不实施除去溶剂所需的成本密集型步骤的情况下进行。如果该经表面反应的碳酸钙以干燥形式提供,则经干燥的经表面反应的碳酸钙具有基于该经表面处理的经表面反应的碳酸钙重量的总重量计为0.001-30%重量的水含量。例如,步骤a)中提供的该经表面反应的碳酸钙具有基于步骤a)中提供的该经表面反应的碳酸钙的干重计为小于15%重量、优选小于10.0%重量的含湿量。在一种实施方案中,步骤a)中提供的该经表面反应的碳酸钙具有基于步骤a)中提供的该经表面反应的碳酸钙的干重计为0.01%重量-10.0%重量、优选0.02%重量-8.0%重量且更优选0.04%重量-7%重量的含湿量。根据一种优选的实施方案,经干燥的经表面反应的碳酸钙为自由流动的粉末。步骤(ii)的表征:提供表面处理剂根据本发明的步骤(ii),提供表面处理剂。更确切的说,提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。另外可选地,提供至少一种水溶性金属盐、水溶性过渡金属络合物、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐、钯盐、铂盐及其混合物。应理解,该表面处理剂可为一种或多种表面处理剂。在本发明的一种实施方案中,该表面处理剂包括一种类型的表面处理剂,优选由一种类型的表面处理剂构成。另外可选地,该表面处理剂包括两种或更多种类型的表面处理剂,优选由两种或更多种类型的表面处理剂构成。例如,该表面处理剂包括两种或三种类型的表面处理剂,优选由两种或三种类型的表面处理剂构成。优选地,该表面处理剂包括一种类型的表面处理剂,更优选由一种类型的表面处理剂构成。如以上已经阐述的,该表面处理剂为至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。根据本发明的水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。根据本发明的术语“水溶性金属盐”被定义为包含金属离子并且是水溶性的盐。该水溶性金属盐可另外可选地选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐、钯盐、铂盐及其混合物。根据本发明的术语“水溶性金属盐”被定义为包含金属离子并且是水溶性的盐。根据本发明的铬盐是包含铬离子、优选cr2+和/或cr3+的盐。例如,该铬盐选自溴化铬(crbr3),氯化铬(crcl2),氟化铬(crf2),硝酸铬(cr(no3)3)和高氯酸铬(cr(clo4)3)。该至少一种水溶性铬盐可为无水盐或水合盐。根据本发明的锰盐为包含锰离子、优选mn2+和/或mn3+的盐。例如,该锰盐选自溴化锰(mnbr2),氯化锰(mncl2),硝酸锰(mn(no3)2)和硫酸锰(mnso4)。该至少一种水溶性锰盐可为无水盐或水合盐。根据本发明的铁盐为包含铁离子、优选fe2+和/或fe3+的盐。例如,该铁盐选自溴化铁(febr2),氯化铁(fecl2、fecl3),碘化铁(fei2),硝酸铁(fe(no3)3),六氰合铁(ⅱ)酸钾(k4fe(cn)6),硫酸亚铁铵(nh4)2fe(so4)2和硫酸亚铁(feso4)。该至少一种水溶性铁盐可为无水盐或水合盐。根据本发明的钴盐为包含钴离子、优选co2+和/或co3+的盐。例如,该钴盐选自溴化钴(cubr2),氯化钴(cocl2),氯酸钴(co(clo3)2),碘化钴(coi2),硝酸钴(co(no3)2)和硫酸钴(coso4)。该至少一种水溶性钴盐可为无水盐或水合盐。根据本发明的铜盐为包含铜离子、优选cu+和/或cu2+的盐。优选地,该至少一种水溶性铜盐为水溶性铜(ii)盐(即铜盐),其中铜处于2价氧化态。例如,该铜盐选自溴化铜(cubr2),氯化铜(cucl2),氟化铜(cuf2),硝酸铜(cu(no3)2),醋酸铜(c4h6cuo4)和硫酸铜(cuso4)。该至少一种水溶性铜盐可为无水盐或水合盐。根据本发明的锌盐为包含锌离子、优选zn2+的盐。例如,该锌盐选自溴化锌(znbr2),氯化锌(zncl2),硝酸锌(zn(no3)2),碘化锌(zni2)和柠檬酸锌((c6h5o7)2zn3)。该至少一种水溶性锌盐可为无水盐或水合盐。根据本发明的银盐为包含银离子、优选ag+和/或ag2+的盐。例如,该银盐选自氟化银(agf),高氯酸银(agclo4)和硝酸银(agno3)。该至少一种水溶性银盐可为无水盐或水合盐。水溶性钯盐的实例为硫酸钯(ii)。该水溶性钯盐可为无水盐或水合盐。另外可选地,该水溶性钯盐可为硝酸钯(ii),四氨合碳酸氢钯,和/或四氨合氯化钯。水溶性铂盐的实例为溴化铂(iv)或氯化铂(iv)。该水溶性铂盐可为无水盐或水合盐。另外可选地,该水溶性铂盐可为六氯铂酸钠(na2ptcl6),六氯铂酸铵((nh4)2ptcl6)或氯铂酸(h2ptcl6)。水溶性过渡金属络合物的实例为na2pdcl4,na2ptcl4,pd(oac)2,pd(h2nch2ch2nh2)cl2,其中oac表示乙酸基。如在本文中所用的“水合物”是含有以确定比例结合的水分子作为晶体组成部分的无机盐。取决于每个盐化学式单元的水分子数目,该水合物可被称为一水合物、二水合物、三水合物、四水合物、五水合物、六水合物、七水合物、八水合物、九水合物、十水合物、半水合物等。根据本发明的“水溶性金属氢氧化物”被定义为包含过渡金属离子且为水溶性的氢氧化物。该水溶性金属氢氧化物可选自本领域已知的任何水溶性过渡金属氢氧化物。根据本发明的“水溶性金属氧化物”被定义为包含过渡金属离子且为水溶性的氧化物。根据本发明的一种实施方案,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为水溶性金属盐,优选选自铜盐、锌盐、银盐及其混合物,甚至更优选为铜盐,选自硝酸铜、硫酸铜、醋酸铜、氯化铜、氟化铜、溴化铜、其水合物及其混合物,且最优选选自硫酸铜、其水合物及其混合物。根据另一实施方案,该至少一种水溶性铜盐选自硝酸铜、六水合硝酸铜、醋酸铜、一水合醋酸铜、氯化铜、二水合氯化铜、氟化铜、硫酸铜、五水合硫酸铜及其混合物。然而,该至少一种水溶性铜盐可选自本领域已知的任何其他水溶性铜盐。该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物可以溶液的形式或作为干燥材料提供。根据一种优选的实施方案,该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物作为干燥材料提供。该干燥材料可为粉末、薄片、粒料等形式,且最优选为粉末形式。步骤(iii)的表征:提供水性溶剂根据本发明的步骤(iii),提供水性溶剂。根据本发明的“水性溶剂”是包含水和任选的与水混溶的其他溶剂的溶液,该其他溶剂例如是有机溶剂如甲醇、乙醇、正丁醇、异丙醇、正丙醇及其混合物。根据一种优选的实施方案,该溶剂由水构成。根据另一种优选的实施方案,该溶剂为水和至少一种与水混溶的有机溶剂的混合物。优选地,该“水性溶剂”包含基于该水性溶剂的总体积计为至少50%体积的水、更优选60%体积的水、甚至更优选70%体积的水且最优选至少80%体积的水。例如,该水性溶剂为由水和乙醇构成的混合物,其中水:乙醇的混合物具有基于溶剂的体积计为1:1的比。根据本发明的一种优选实施方案,步骤(iii)的该水性溶剂由水构成。步骤(iv)的表征:含金属的溶液的制备根据本发明的步骤(iv),使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液。根据本发明的“含金属的溶液”是包含水和任选的与水混溶的其他溶剂以及金属离子的溶液。金属离子从步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物获得。该“含金属的溶液”可包含一种或多种来源的金属离子,例如两种或三种来源的金属离子。根据本发明的一种实施方案,该含金属的溶液仅由水、金属离子和抗衡离子构成且并不含有其他与水混溶的溶剂。根据本发明的另一种实施方案,该含金属的溶液仅包含来自一种来源的金属离子。优选地,该含金属的溶液只由水、一种类型的金属离子和一种类型的抗衡离子构成。例如,该含金属的溶液包含水和硫酸铜或者该含金属的溶液由水和硫酸铜构成。为了制备含金属的溶液的步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂的接触可通过本领域技术人员已知的任何传统手段实现。优选地,该接触可在混合和/或均质化和/或溶解的条件下进行。本领域技术人员将根据其方法设备调适这些混合和/或均质化和/或溶解的条件如混合速度、时间和温度。例如,该混合和均质化可借助于犁铧式(ploughshare)混合器发生。犁铧式混合器以机械方式产生的流化床为原理进行工作。犁铧式叶片靠近水平圆柱形转鼓的内壁旋转并且输送混合物的组分离开产品床并进入到敞开的混合空间中。机械方式产生的流化床确保在非常短时间内即使是大批量产品的强烈混合。切碎机和/或分散器用于在干燥操作中分散结块。可用于本发明的设备可例如从德国的gebrüdermaschinenbaugmbh或从德国的viscojetrührsystemegmbh获得。在进行接触步骤(iv)步骤之前,可将步骤(iii)的该水性溶剂预加热。例如,在进行接触步骤(iv)之前,可将步骤(iii)的该水溶液在20-100℃、优选25-90℃、更优选30-60℃且最优选35-50℃的温度下预加热。例如,可将步骤(iii)的该水性溶剂加热至40℃±5℃的温度。另外可选地,可将步骤(iii)的该水性溶剂加热至25℃±5℃的温度。该加热可在精确地为100000pa(1巴,14.5psi,0.98692atm)的绝对压力下进行。任选地,该加热在减压或升高的压力(即在低于或高于100000pa的压力下)下进行。用于进行步骤(iii)的该水性溶剂的预加热的处理时间为120min或更短的时间周期,优选60分钟或更短的时间周期,且最优选30分钟或更短的时间周期。步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂的接触步骤可在一个或多个步骤中进行。例如,该方法可为连续方法。在该情况下,可以恒流方式添加步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,使得在步骤(iv)期间提供步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物的恒定浓度。另外可选地,在一个步骤中向步骤(iii)的水溶液中添加步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物。在另一种实施方案中,本发明的方法可为间歇方法,即在不止一个步骤中使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)获得的水溶液接触。另外可选地,也可将步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物以非等份(即以较大或较小份)添加到步骤(iii)的该水性溶剂中。根据本发明的一种实施方案,步骤c)以间歇或连续方法进行0.1秒-1000秒的时间周期。例如,步骤(iv)为连续方法且包括一个或多个接触步骤并且总接触时间为0.1-20秒、优选0.5-15秒且最优选1-10秒。根据本发明的一种实施方案,步骤(iv)中获得的该含金属的溶液具有基于该金属盐溶液的总重量计为1-80%重量、优选5-60%重量且最优选10-50%重量的固体含量。步骤(iv)中获得的该含金属的溶液可被搅拌1分钟-4小时的时间周期。例如,步骤(iv)中获得的该含金属的溶液可被搅拌10分钟、30分钟、45分钟、1小时、2小时或3小时的时间周期。可将步骤(iv)中获得的该含金属的溶液过滤。过滤方法可为本领域技术人员已知的任何方法,例如,在布氏漏斗中通过滤纸过滤。该过滤可在精确地为100000pa(1巴,14.5psi,0.98692atm)的绝对压力下进行。任选地,加热在减压或升高的压力下进行,即在低于或高于100000pa的压力下进行。步骤(v)的表征:接触步骤以获得混合物根据本发明的步骤(v),在一个或多个步骤中使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物。该接触可在一个或多个步骤中进行以形成混合物。根据本发明的一种实施方案,步骤(v)包括如下步骤:在第一步骤中提供步骤(i)的该至少一种经表面反应的碳酸钙,并且随后添加步骤(iv)的该含金属的溶液。根据本发明的另一实施方案,步骤(v)包括如下步骤:在第一步骤中提供步骤(iv)的该含金属的溶液,并且随后添加步骤(i)的该至少一种经表面反应的碳酸钙。根据又另一种实施方案,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液同时接触。根据一种优选的实施方案,步骤(v)包括如下步骤:在第一步骤中提供步骤(i)的该至少一种经表面反应的碳酸钙,并且随后添加步骤(iv)的该含金属的溶液。可以恒流方式添加步骤(iv)的该含金属的溶液。另外可选地,可在一个步骤中将步骤(iv)的该含金属的溶液添加到步骤(i)的该至少一种经表面反应的碳酸钙中。也可在不止一个步骤中将步骤(iv)的该含金属的溶液添加到步骤(i)的该至少一种经表面反应的碳酸钙中。另外可选地,也可将步骤(iv)的该含金属的溶液以非等份(即以较大或较小份)添加到步骤(i)的该至少一种经表面反应的碳酸钙中到步骤(iii)的水性溶液中。步骤(i)的该经表面反应的碳酸钙可以干燥形式、优选以粉末形式提供或可以悬浮液的形式、优选以水性悬浮液的形式提供,其中该经表面反应的碳酸钙的固体含量基于该水性悬浮液的总重量计在1-80%重量、更优选3-70%重量,且甚至更优选5-60%重量的范围内。根据一种优选的实施方案,步骤(i)的该经表面反应的碳酸钙以悬浮液的形式、优选以水性悬浮液的形式提供,其中该经表面反应的碳酸钙的固体含量基于该水性悬浮液的总重量计在1-80%重量、更优选3-70%重量,且甚至更优选5-60%重量的范围内。根据一种示例性的实施方案,步骤(v)包括如下步骤:在第一步骤中以水性悬浮液的形式提供步骤(i)的该至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙的固体含量基于该水性悬浮液的总重量计在1-80%重量的范围内;并且随后添加步骤(iv)的该含金属的溶液。根据另一种实施方案,步骤(v)由以下操作构成:在一个或多个步骤中使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物。该接触步骤(v)可通过本领域已知的任何手段进行。例如,可通过喷雾和/或混合使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触。用于喷雾或混合的适合的方法设备是本领域技术人员已知的。根据本发明的一种实施方案,方法步骤(v)通过喷雾进行。根据本发明的另一实施方案,方法步骤(v)通过混合进行。步骤(v)中的混合可通过本领域技术人员已知的任何传统手段实现。本领域技术人员将根据其方法设备调适混合条件如混合速度、分割以及温度。额外地,可在均质化和/或粒子分割的条件下进行混合。例如,该混合和均质化可借助于犁铧式混合器发生。犁铧式混合器以机械方式产生的流化床为原理进行工作。犁铧式叶片靠近水平圆柱形转鼓的内壁旋转并且输送混合物的组分离开产品床并进入到敞开的混合空间中。机械方式产生的流化床确保在非常短的时间内即使是大批量产品的强烈混合。切碎机和/或分散器用于在干燥操作中分散结块。可用于本发明的设备可例如从德国的gebrüdermaschinenbaugmbh或从德国的viscojetrührsystemegmbh获得。根据本发明的另一种实施方案,步骤(v)进行至少1秒、优选至少1分钟,例如至少10分钟、15分钟、30分钟、45分钟或60分钟。根据一种优选的实施方案,步骤d)进行1秒-60分钟的时间周期,优选15分钟-45分钟的时间周期。例如,混合步骤d)进行30分钟±5分钟。根据本发明的一种优选的实施方案,步骤d)在30-120℃的温度下和/或1秒-60分钟的时间周期内进行。也在本发明的范围中的是:可在方法步骤(v)期间引入额外的水,例如以用于控制和/或维持和/或实现获得的混合物所希望的固体含量或布氏粘度。根据一种实施方案,步骤(v)中获得的混合物的固体含量基于该混合物的总重量计为0.5-80%重量、优选1-70%重量。获得的混合物的布氏粘度可为10-10000mpa·s、优选100-2000mpa·s。根据本发明的一种实施方案,该至少一种金属盐、金属氢氧化物、金属氧化物或其混合物以基于该至少一种经表面反应的碳酸钙的总干重计为0.001-80%重量、优选0.01-60%重量、更优选0.05-50%重量且最优选1-40%重量的量添加到该至少一种经表面反应的碳酸钙中。根据本发明的一种实施方案,在步骤(v)之前和/或期间将呈水溶液或水性悬浮液形式的碱添加到步骤(i)的该至少一种经表面反应的碳酸钙中。在步骤(v)之前和/或期间添加的碱优选包含碳酸根离子,并且可例如选自碳酸钠、碳酸钾、碳酸钡、碳酸锰及其混合物,且最优选该碱为碳酸钠。根据本发明的一种优选实施方案,在步骤(v)之前将碱添加到步骤(i)的该经表面反应的碳酸钙中。该碱可以基于该至少一种经表面反应的碳酸钙的总干重计为0.001-80%重量、优选0.01-60%重量、更优选0.05-50%重量且最优选1-40%重量的量添加。例如,该碱以基于该至少一种经表面反应的碳酸钙的总干重计为10-50%重量的量添加。该碱可以固体形式或作为“液体”添加。例如,碳酸钠、碳酸钾、碳酸钡、碳酸锰在标准环境温度和压力(satp)下是固体,该标准环境温度和压力(satp)是指298.15k(25℃)的温度以及精确地为100000pa(1巴,14.5psi,0.98692atm)的绝对压力。如果该碱为固体形式,则其可在步骤(v)之前和/或期间添加,例如作为粉末、片剂、粒料、薄片等添加。然而,在步骤(v)之前和/或期间,固体碱也可分散/悬浮在水中并且作为分散体/悬浮液(即以液体形式)添加。步骤(vi)的表征:混合物的加热根据本发明的步骤(vi),将从步骤(v)获得的该混合物加热至20-250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。在本发明方法的步骤(vi)中,将步骤(v)的该混合物加热至20-250℃的温度以形成经表面处理的经表面反应的碳酸钙。根据一种实施方案,在步骤(vi)中,将从步骤(v)获得的该混合物加热至50-180℃、优选60-160℃、甚至更优选80-150℃且最优选90-130℃的温度。步骤(v)和步骤(vi)的温度可相同或不同。根据一种优选的实施方案,步骤(v)和步骤(vi)的温度相同。在本发明的主旨内,加热意味着将从步骤(v)获得的该混合物在20-250℃范围的温度下或在优选的温度范围之一的温度下保持一段有限的时间周期。术语“加热”并不将根据本发明的方法局限为如下这样的过程:其中在步骤(vi)期间通过经由外部热源添加能量将混合物的温度调节至20-250℃的温度范围,而是还包括保持预加热的混合物,该预加热的混合物通过例如提供步骤(i)的预加热的至少一种经表面反应的碳酸钙和/或步骤(iv)的预加热的含金属的溶液或在步骤(v)期间预加热混合物来获得。为完整起见,应当提及,在从步骤(v)获得的该混合物的温度高于250℃的情况下,将温度调节至20-250℃的范围也被该术语加热所涵盖。根据本发明的一种实施方案,在本发明方法的步骤(v)中,将步骤(v)的该混合物调节至20-250℃的温度以形成经表面处理的经表面反应的碳酸钙。在本发明的方法以连续方法的形式进行的情况下,该加热步骤可进行3秒-60秒,且优选10秒-30秒。在本发明的方法以间歇方法的形式进行的情况下,该加热步骤可进行1-60分钟、优选3-40分钟且更优选5-30分钟。另外可选地,该加热步骤可进行比60分钟更长的时间周期,例如2小时或3小时或4小时。该加热步骤不需要超过5小时。本领域技术人员将理解,步骤(vi)在足以形成经表面处理的经表面反应的碳酸钙的时间周期内进行。根据本发明的一种实施方案,方法步骤(vi)进行至少1分钟、优选至少5分钟。根据一种实施方案,方法步骤(vi)通过将混合物加热至20-250℃的温度至少10分钟、优选至少20分钟、更优选至少1小时、甚至更优选至少2小时且最优选至少3小时来进行。根据一种实施方案,进行该加热步骤(vi),直到10-99.8%摩尔、优选20-95%摩尔的步骤(ii)中提供的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物在该至少一种经表面反应的碳酸钙的表面的至少一部分上以水不溶性金属化合物的形式沉淀以形成经表面处理的经表面反应的碳酸钙,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。据信,不溶性金属化合物的沉淀遵循阿仑尼乌斯方程(arrheniusequation),其是指升高温度能缩短反应时间并且降低温度导致延长的反应时间。加热步骤(vi)可在减压、环境压力下或在升高的压力下进行。对于高于100℃的温度,优选在升高的压力下进行加热步骤。也在本发明的范围内的是:方法步骤(v)和方法步骤(vi)可同时进行。因而,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触的步骤以及将混合物加热至20-250℃的加热步骤可同时进行。借助于步骤(vi),获得经表面处理的经表面反应的碳酸钙。根据一种实施方案,该经表面处理的经表面反应的碳酸钙以水性悬浮液的形式获得。该水性悬浮液可具有基于该悬浮液中的该经表面处理的经表面反应的碳酸钙的重量计为0.8-80%重量、更优选1-50%重量的固体含量。如果必要的话,在方法步骤(vi)期间可引入额外的水,例如,为了控制和/或维持和/或实现获得的水性悬浮液所希望的固体含量或布氏粘度。本发明人令人惊讶地发现,通过前述方法可以制备具有改善特性的经表面处理的经表面反应的碳酸钙。更确切的说,通过使用本发明的方法,可以制备包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。因此,与其中表面处理剂与经表面反应的碳酸钙的反应产物仅被吸收进微粒状材料的孔中或涂覆或吸附在微粒状材料的内或外表面上的传统微粒状材料相比,从表面处理剂(即至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物)与经表面反应的碳酸钙获得的反应产物更牢固地附着到经表面反应的碳酸钙的表面上。归因于这种固定化和/或牢固的附着,形成的该至少一种水不溶性金属化合物不能用水从该经表面反应的碳酸钙的表面上洗掉。在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成的该至少一种水不溶性金属化合物可通过本领域技术人员已知的任何方法检测或分析。例如,在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物可通过xrd,例如用brukeradvanced8粉末衍射仪来检测。根据本发明的一种实施方案,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为硫酸铜、其水合物或其混合物和/或在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物选自水合磷酸氢铜(cuhpo4·h2o),孔雀石(cu2co3(oh)2),水胆矾(cu4so4(oh)6),钙铜矾(cacu4(so4)2(oh)6·3h2o),一水蓝铜矾(cu4(so4)(oh)6·h2o)及其混合物。另外可选地,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为硫酸锌、其水合物或其混合物和/或在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物为磷酸氢锌(znh2p2o7)。另外可选地,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为氯化锌、其水合物或其混合物和/或在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物为水合磷酸锌(zn3(po4)2·h2o)。根据本发明的另一种实施方案,步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为硫酸锌和/或氯化锌、其水合物或其混合物。根据本发明的另一种实施方案,在该经表面反应的碳酸钙的表面上形成的该至少一种水不溶性金属化合物选自磷酸铬(crpo4),碳酸铬(cr2(co3)),磷酸锰(mn3(po4)2),碳酸锰(mnco3),磷酸铁(iii)(fepo4),磷酸铁(ii)(fe3(po4)2),水合磷酸氢铁(fe3h9(po4)6·6h2o,菱铁矿(feco3),磷酸钴(co3(po4)2),磷酸氢钴(co2(hpo4)3,碳酸钴(coco3),磷酸银(ag3po4),磷酸氢银(ag2hpo4),碳酸银(ag2co3)及其混合物。额外的方法步骤根据一种实施方案,本发明的方法进一步包括在步骤(vi)之前、期间或之后将从步骤(v)获得的该混合物研磨和/或分级和/或分类的步骤。根据另一实施方案,该方法进一步包括在步骤(vi)之前过滤从步骤(v)获得的该混合物的步骤。根据又另一种实施方案,该方法进一步包括如下的步骤:在步骤(vi)之前将从步骤(v)获得的该混合物研磨和/或分级和/或分类,并且随后过滤所获得的研磨的混合物。该研磨步骤可通过本领域技术人员熟知的用于湿法研磨的所有技术和研磨机进行。该研磨步骤可例如在使得粉碎主要由使用辅助体冲击产生的条件下,用传统研磨装置进行,也即在以下的一种或多种中进行:球磨机、棒磨机、振动研磨机、离心冲击研磨机、立式珠磨机、磨碎机或本领域技术人员已知的其他此类设备。该研磨步骤可以间歇或连续方式、优选连续方式进行。优选地,该研磨步骤可在不添加与如步骤(i)和(ii)中所提供的化合物不同的化合物的情况下进行。可将从步骤(v)获得的该混合物过滤以除去盐或水。获得的经表面处理的经表面反应的碳酸钙可进行进一步加工,例如,在以悬浮液的形式获得该经表面处理的经表面反应的碳酸钙的情况下,该经表面处理的经表面反应的碳酸钙可从水性悬浮液分离和/或经历洗涤步骤和/或表面处理步骤和/或干燥步骤。根据本发明的一种实施方案,在本发明方法的步骤(v)中形成的混合物为水性悬浮液并且该方法进一步包括步骤(vii):在步骤(vi)之后,从该水性悬浮液分离该经表面处理的经表面反应的碳酸钙。因而,制造经改性的矿物基填料的方法可包括以下步骤:(i)提供至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应,(ii)提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物,(iii)提供水性溶剂,(iv)使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液,(v)在一个或多个步骤中,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物,以及(vi)加热从步骤(v)获得的该混合物至20–250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成,以及(vii)从由步骤(vi)获得的水性悬浮液分离该经表面处理的经表面反应的碳酸钙。来自步骤(vi)的该经表面处理的经表面反应的碳酸钙可通过本领域技术人员已知的任何传统的分离手段从水性悬浮液分离。根据本发明的一种实施方案,本发明的方法进一步包括步骤(x):在加热步骤(vi)之前将从步骤(v)获得的该混合物优选通过离心或过滤进行机械脱水。该机械脱水过程的实例为过滤(例如借助于转鼓过滤机或压滤机)、纳滤或离心。另外可选地,脱水可以热方式进行。热脱水过程的实例为通过施加热的浓缩过程(例如在蒸发器中)。根据一种优选的实施方案,机械脱水通过过滤和/或离心进行。根据本发明的一种实施方案,该方法进一步包括步骤(viii):用水洗涤从步骤(vi)或如果存在的步骤(vii)获得的该经表面处理的经表面反应的碳酸钙。该经表面处理的经表面反应的碳酸钙可用水和/或适合的溶剂(优选水)洗涤。适合的溶剂是本领域已知的,例如为具有4-14个碳原子的脂族醇、醚和二醚,二醇、烷氧基化二醇、二醇醚、烷氧基化芳族醇、芳族醇、其混合物或其与水的混合物。例如,该经表面处理的经表面反应的碳酸钙可用水和/或适合的溶剂(优选水)洗涤一次、两次或三次。在分离之后,可干燥该经表面处理的经表面反应的碳酸钙以获得经干燥的经表面处理的经表面反应的碳酸钙。根据一种实施方案,本发明的方法进一步包括步骤(ix):在步骤(vi)或如果存在的步骤(vii)或步骤(viii)之后在60-600℃、优选80-550℃的温度下干燥经表面处理的经表面反应的碳酸钙。优选地,进行干燥,直到经表面处理的经表面反应的碳酸钙的含湿量基于经干燥的经改性的矿物基填料的总重量计为0.01-5%重量为止。通常,干燥步骤(ix)可使用任何适合的干燥设备进行,并且可例如包括热干燥和/或减压下干燥(使用设备如蒸发器、闪蒸干燥器、烘箱、喷雾干燥器)和/或在真空室中干燥。干燥步骤(ix)可在减压下、环境压力下或在升高的压力下进行。对于低于100℃的温度,可优选在减压下进行干燥步骤。根据一种优选的实施方案,该分离通过热方法进行。这可允许在不改变设备的情况下随后干燥该经改性的矿物基填料。根据本发明的一种实施方案,在接触步骤(v)之前或期间,将步骤(i)的该经表面反应的碳酸钙加热至20-200℃、优选30-160℃、甚至更优选50–150℃且最优选80–130℃的温度。额外的表面处理在本发明方法的步骤(vi)中形成的该经表面处理的经表面反应的碳酸钙可进行后处理,优选在如果存在的步骤(vii)、(viii)、(ix)或(x)之后进行。然而,也可以在湿产品上进行额外的表面处理。根据一种实施方案,用脂肪酸如硬脂酸、硅烷、或脂肪酸的磷酸酯、或硅氧烷处理该经表面处理的经表面反应的碳酸钙。根据一种实施方案,本发明的方法进一步包括如下的步骤:在步骤(vi)期间和/或之后在一个或多个步骤中用至少一种疏水剂、优选在30-200℃的温度下处理在步骤(vi)中形成的该经表面处理的经表面反应的碳酸钙。优选地,额外的表面处理在经干燥的经表面处理的经表面反应的碳酸钙上进行。因而,根据本发明的一种实施方案,本发明的方法包括步骤(ix):干燥该经表面处理的经表面反应的碳酸钙,并且在干燥步骤(ix)之后在一个或多个步骤中用至少一种疏水剂、优选在30-200℃的温度下处理该经干燥的经表面处理的经表面反应的碳酸钙。根据一种实施方案,在步骤(vi)期间和/或之后在一个或多个步骤中用至少一种疏水剂、在30-200℃、优选60-130℃、更优选70-120℃且最优选80-110℃的温度下处理在步骤(vi)中形成的该经表面处理的经表面反应的碳酸钙。根据一种实施方案,该至少一种疏水剂的添加量使得该至少一种疏水剂在该经表面处理的经表面反应的碳酸钙的总表面积上的总重量为0.001-10mg/m2、优选0.001-9mg/m2、更优选0.01-8mg/m2且最优选0.1-4mg/m2。根据本发明的一种实施方案,该方法进一步包括如下的步骤:在步骤(vi)期间和/或之后在一个或多个步骤中用至少一种疏水剂、在30-200℃的温度下处理在步骤(vi)中形成的该经表面处理的经表面反应的碳酸钙,其中该疏水剂的加入量使得该至少一种疏水剂在该经表面处理的经表面反应的碳酸钙的总表面积上的总重量为0.001-10mg/m2。适合的疏水剂为例如脂肪酸、脂族羧酸、脂族羧酸酯、单取代琥珀酸酐、单取代琥珀酸或磷酸酯。适合的疏水剂以及其用于制备经表面处理的填料产品的方法例如描述在以下文献中:ep2159258a1、ep2390285a1、ep2390280a1、wo2014/060286a1和wo2014/128087a1。在一种实施方案中,该疏水剂为具有c4-c24的碳原子总数的脂族羧酸和/或其反应产物。在本发明的含义中,术语脂族羧酸的“反应产物”是指通过使经改性的矿物基填料与该至少一种脂族羧酸接触而获得的产物。所述反应产物在该至少一种脂族羧酸的至少一部分与位于该经表面处理的经表面反应的碳酸钙粒子的表面处的反应性分子之间形成。在本发明含义中的脂族羧酸可选自一种或多种直链、支化链、饱和、不饱和和/或脂环族羧酸。优选地,该脂族羧酸为一元羧酸,即该脂族羧酸的特征在于存在单个羧基。所述羧基位于碳骨架的末端处。在本发明的一种实施方案中,该脂族羧酸选自饱和非支化的羧酸,也即该脂族羧酸优选选自以下的羧酸:戊酸、己酸、庚酸、辛酸、壬酸、癸酸、十一烷酸、月桂酸、十三烷酸、肉豆蔻酸、十五烷酸、棕榈酸、十七烷酸、硬脂酸、十九烷酸、花生酸、二十一烷酸(heneicosylicacid)、山萮酸、三十烷酸、木蜡酸及其混合物。在本发明的另一种实施方案中,该脂族羧酸选自辛酸、癸酸、月桂酸、肉豆蔻酸、棕榈酸、硬脂酸、花生酸及其混合物。优选地,该脂族羧酸选自肉豆蔻酸、棕榈酸、硬脂酸及其混合物。例如,该脂族羧酸是硬脂酸。额外地或另外可选地,该疏水剂可为至少一种单取代的琥珀酸和/或盐性反应产物和/或一种或多种磷酸单酯和/或其反应产物与一种或多种磷酸二酯和/或其反应产物的至少一种磷酸酯掺合物。用这些疏水剂处理含碳酸钙材料的方法例如描述在ep2722368a1和ep2770017a1中。根据一种实施方案,该至少一种疏水剂选自具有c4-c24的碳原子总数的脂族羧酸和/或其反应产物,由利用选自在取代基中的碳原子总数为c2至c30的线性、支化、脂族和环状基团的基团单取代的琥珀酸酐所构成的单取代琥珀酸酐和/或其反应产物,一种或多种磷酸单酯和/或其反应产物与一种或多种磷酸二酯和/或其反应产物的磷酸酯掺合物,聚氢硅氧烷及其反应产物,惰性有机硅油(优选聚二甲基硅氧烷),及其混合物。术语“琥珀酸酐”(也称为二氢-2,5-呋喃二酮、琥珀酸的酸酐或琥珀酰化氧)具有分子式c4h4o3并且为琥珀酸的酸酐。在本发明含义中的术语“单取代琥珀酸酐”是指其中氢原子被另一取代基取代的琥珀酸酐。在本发明含义中的术语“至少一种单取代琥珀酸酐的反应产物”是指通过使经表面处理的经表面反应的碳酸钙与一种或多种单取代琥珀酸酐接触而获得的产物。所述盐性反应产物在由应用的单取代琥珀酸酐所形成的单取代琥珀酸与位于该经表面处理的经表面反应的碳酸钙的表面处的反应性分子之间形成。在本发明含义中的术语“磷酸单酯”是指利用一个醇分子单酯化的正磷酸分子,所述醇分子选自在醇取代基中具有碳原子总量为c6至c30、优选c8至c22、更优选c8至c20并且最优选c8至c18的不饱和或饱和、支化或线性、脂族或芳族的醇。在本发明含义中的术语“磷酸二酯”是指利用两个醇分子二酯化的正磷酸分子,所述醇分子选自在醇取代基中具有碳原子总量为c6至c30、优选c8至c22、更优选c8至c20并且最优选c8至c18的相同或不同的不饱和或饱和、支化或线性、脂族或芳族的醇。在本发明含义中的术语“磷酸酯或一种或多种磷酸单酯和/或一种或多种磷酸二酯的掺合物的盐性反应产物”是指通过含碱土金属碳酸盐材料与一种或多种磷酸单酯和一种或多种磷酸二酯和任选的磷酸接触所获得的产物。所述盐性反应产物在应用的一种或多种磷酸单酯和一种或多种磷酸二酯和任选的磷酸与位于该经表面处理的经表面反应的碳酸钙的表面处的反应性分子之间形成。经表面处理的经表面反应的碳酸钙根据本发明,提供通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙。根据本发明的另一种实施方案,提供通过包括以下步骤的方法可获得的经表面处理的经表面反应的碳酸钙:(i)提供至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应,(ii)提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物,(iii)提供水性溶剂,(iv)使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液,(v)在一个或多个步骤中,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物,以及(vi)加热从步骤(v)获得的该混合物至20-250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。例如,通过使用本发明的方法,可以制备具有改善特性的经表面处理的经表面反应的碳酸钙。更确切的说,通过使用本发明的方法,可以制备包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。因此,与其中表面处理剂仅被吸收或涂覆在微粒状材料的表面上的传统微粒状材料相比,该表面处理剂(也即该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物)更牢固地附着到该经表面反应的碳酸钙的表面上。归因于这种更牢固的附着,所形成的该至少一种水不溶性金属化合物不能用水从该经表面反应的碳酸钙的表面洗掉。这可例如通过如下方式来分析:洗涤该经表面处理的经表面反应的碳酸钙并且用icp-oes分析洗涤水。这种操作程序是本领域技术人员已知的并且被描述在实施例部分中。本发明的经表面处理的经表面反应的碳酸钙可以经表面处理的经表面反应的碳酸钙的悬浮液的形式、作为经分离的经表面处理的经表面反应的碳酸钙或作为经干燥的经表面处理的经表面反应的碳酸钙的形式提供。根据一种优选的实施方案,该经表面处理的经表面反应的碳酸钙为经干燥的(优选为粉末形式)的经表面处理的经表面反应的碳酸钙。在该经表面处理的经表面反应的碳酸钙已经被干燥的情况下,经干燥的经表面处理的经表面反应的碳酸钙的含湿量基于该经干燥的经表面处理的经表面反应的碳酸钙的总重量计为0.01-30%重量。例如,该经干燥的经表面处理的经表面反应的碳酸钙的含湿量基于该经干燥的经表面处理的经表面反应的碳酸钙的总重量计为小于或等于20.0%重量、优选小于10.0%重量、甚至更优选小于5.0%重量,根据一种实施方案,该经干燥的经表面处理的经表面反应的碳酸钙的含湿量基于该经干燥的经表面处理的经表面反应的碳酸钙的总重量计为小于或等于1.0%重量,优选小于或等于0.5%重量,且更优选小于或等于0.2%重量。根据另一实施方案,该经干燥的经表面处理的经表面反应的碳酸钙的含湿量基于该经干燥的经表面处理的经表面反应的碳酸钙的总重量计为0.01-0.15%重量、优选0.02-0.10%重量且更优选0.03-0.07%重量。根据本发明的经表面处理的经表面反应的碳酸钙可具有(a)根据iso9277使用氮气和bet方法测量的15m2/g-200m2/g、优选27m2/g-180m2/g、更优选25m2/g-160m2/g且最优选30m2/g-150m2/g的比表面积,和/或(b)1-75μm、优选1.3-50μm、更优选1.5-40μm、甚至更优选1.8-30μm且最优选2-15μm的体积中值颗粒直径d50(体积),和/或(c)2-150μm、优选4-100μm、更优选6-80μm、甚至更优选8-60μm且最优选10-30μm的颗粒直径d98(体积),和/或(d)由汞孔隙率测定法测量结果计算的0.1-2.3cm3/g、更优选0.2-2.0cm3/g、尤其优选0.4-1.8cm3/g且最优选0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的经表面处理的经表面反应的碳酸钙可具有(a)根据iso9277使用氮气和bet方法测量的30m2/g-150m2/g的比表面积,以及(b)2-15μm的体积中值颗粒直径d50(体积),以及任选地(c)10-30μm的颗粒直径d98(体积),以及任选地(d)由汞孔隙率测定法测量结果计算的0.6-1.6cm3/g的粒子内侵入式比孔容。根据本发明的一种实施方案,该经表面处理的经表面反应的碳酸钙包含至少一种选自以下的化合物作为在该经表面反应的碳酸钙的表面上形成的水不溶性金属化合物:水合磷酸氢铜(cuhpo4·h2o),孔雀石(cu2co3(oh)2),水胆矾(cu4so4(oh)6),钙铜矾(cacu4(so4)2(oh)6·3h2o),一水蓝铜矾(cu4(so4)(oh)6·h2o)及其混合物。根据本发明的一种实施方案,该经表面处理的经表面反应的碳酸钙包含在该经表面反应的碳酸钙的表面上的不溶性锌盐。本发明的经表面处理的经表面反应的碳酸钙也可以组合物的形式提供和/或使用。根据本发明的一个方面,提供包含根据本发明的经表面处理的经表面反应的碳酸钙的组合物。该组合物可以基于该组合物的总重量计为至少0.5%重量、优选至少2%重量、更优选至少5%重量、甚至更优选至少10%重量且最优选至少20%重量的量包含根据本发明的经表面处理的经表面反应的碳酸钙。至少一种经表面反应的碳酸钙用于固定(immobilization)的用途根据本发明,至少一种经表面反应的碳酸钙用于通过根据本发明的方法制备至少一种经表面处理的经表面反应的碳酸钙而在该至少一种经表面反应的碳酸钙的表面上固定至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应。根据本发明的一种实施方案,至少一种经表面反应的碳酸钙用于通过根据包括以下步骤的方法制备至少一种经表面处理的经表面反应的碳酸钙而在该至少一种经表面反应的碳酸钙的表面上固定至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应:(i)提供至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应,(ii)提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物,(iii)提供水性溶剂,(iv)使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液,(v)在一个或多个步骤中,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物,并且(vi)加热从步骤(v)获得的该混合物至20-250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成。根据本发明的术语“固定(immobilizing)”意味着该至少一种水溶性的水溶性金属盐、水溶性金属氢氧化物和/或水溶性金属氧化物能够在至少一种经表面反应的碳酸钙的存在下在其表面上形成水不溶性盐。因此,与其中表面处理剂与经表面反应的碳酸钙的反应产物仅被吸收到微粒状材料的孔中或涂覆或吸附在微粒状材料的内或外表面上的传统微粒状材料相比,可以将该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物牢固地附着到微粒状材料的表面上,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物。经表面处理的经表面反应的碳酸钙的用途本发明的发明人发现,经表面处理的经表面反应的碳酸钙可在干燥产品或湿产品、优选干燥产品中表现出抗微生物活性。因此,本发明的经表面处理的经表面反应的碳酸钙可用在旨在用于制备干燥或湿产品(其中优选干燥产品)的矿物、填料或颜料的悬浮液、分散体或浆料中,它们被典型地用在纸、油漆、橡胶和塑料工业中如用于造纸的涂料、填料、增量剂和颜料以及水性清漆和油漆。本发明的经表面处理的经表面反应的碳酸钙可完全或部分取代传统的填料。由于表面处理剂被固定在该经表面反应的碳酸钙的表面上并且因此更牢固地附着到该经表面处理的经表面反应的碳酸钙的表面上,因此通过本发明的经表面处理的经表面反应的碳酸钙可提供持久的抗微生物效果,即使是其与水接触,例如在制造水性悬浮液形式的产品期间使用时。此外,本发明的经表面处理的经表面反应的碳酸钙甚至可用在涉及与水或水性液体接触或定期经历水洗涤的制品如油漆或布中。该经表面处理的经表面反应的碳酸钙可用于各种应用。根据一种实施方案,通过根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙或包含其的组合物被用在聚合物应用、纸涂层应用、造纸、油漆、涂料、密封剂、印刷油墨、粘合剂、食品、饲料、药物、混凝土、水泥、化妆品、水处理、工程木材应用、石膏板应用、包装应用和/或农业应用中。工程木材应用可包括用于工程木材产品如木材复合材料,优选中等密度纤维板或刨花板。根据另一实施方案,通过根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙或包含其的组合物被用作防腐剂用于控制气味和/或用于增强和/或介入基底的抗微生物活性。优选地,改性基填料可被用作经干燥的经表面处理的经表面反应的碳酸钙。防腐剂是如下这样的化合物,其保护干燥和/或湿的基底免受归因于微生物作用的腐败和/或降解和/或破坏和/或污损和/或可见的缺陷,和/或防止在基底上和/或基底中微生物的生长和/或防止基底通过微生物的污染和/或防止基底上微生物的定居。根据一种优选的实施方案,该防腐剂作为干燥膜防腐剂起作用。该基底优选为固体状态,如纸表面、木材表面、墙、包装材料的表面或聚合物制品的表面,但也可为湿状态如在水性悬浮液中。根据本发明的“气味”通常被定义为人类或其他动物通过嗅觉感知的通常在非常低的浓度下的一种或多种挥发的化合物。因此,“有气味的物质”是具有嗅味或气味的化合物,即挥发性足以输送至鼻子上部的嗅觉系统中。根据本发明优选待控制的气味是引起令人不愉快感觉的气味即恶臭,但是并不局限于此。这种气味可源自有气味的物质,其优选选自在人类和动物体液和分泌物如月经、血液、血浆、腐脓液;阴道分泌物、粘液、乳汁、尿;粪便;呕吐物和汗液中含有的有气味的物质;源自诸如人或动物组织的腐败物;食品如乳制品、肉和鱼以及水果(如榴莲水果)的有气味的物质。根据本发明的一种优选的实施方案,该有气味的物质选自胺如三乙胺,二乙胺,三甲胺,二氨基丁烷,四亚甲基二胺,五亚甲基二胺,吡啶,吲哚,3-甲基吲哚;羧酸如丙酸,丁酸,3-甲基丁酸,2-甲基丙酸,己酸;硫有机化合物如硫醇如甲硫醇,磷有机化合物如甲膦,二甲膦,其衍生物及其混合物;优选地,该有气味的物质为胺,且最优选地,该有气味的物质为二乙胺。根据本发明的一种示例性的实施方案,该有气味的物质为二乙胺或硫醇如2-丙硫醇。该经表面处理的经表面反应的碳酸钙也可用于增强和/或介入基底的抗微生物活性,所述基底例如是纸张、纸板、聚合物材料、油漆、木材表面、混凝土或植物。根据一种优选的实施方案,该抗微生物活性针对至少一种细菌菌株和/或至少一种霉菌菌株和/或至少一种酵母菌株和/或至少一种藻类。化合物的抗微生物活性是指在所述化合物的存在下显见的微生物生长减少和/或存活微生物的减少。表述“增强抗微生物活性”是指含有该经表面处理的经表面反应的碳酸钙的基底的抗微生物活性比相对于不含所述填料的基底的抗微生物活性高。表述“用于介入基底的抗微生物活性”是指在没有本发明的经表面处理的经表面反应的碳酸钙的情况下在基底中没有显见抗微生物活性。根据一种实施方案,该基底为纸、纸板、聚合物材料、油漆、木材表面、混凝土或植物。根据一种实施方案,聚合物材料为聚合物膜。在本发明含义中的“膜”是相对于其长度和宽度而言中值厚度小的材料的片或层。例如,术语“膜”可指具有小于200μm但大于1μm的中值厚度的材料的片或层。根据一种实施方案,该至少一种细菌菌株选自埃希氏菌属(escherichiasp.)、葡萄球菌属(staphylococcussp.)、栖热菌属(thermussp.)、丙酸杆菌属(propionibacteriumsp.)、红球菌属(rhodococcussp.)、土著杆菌属(panninobactersp.)、柄杆菌属(caulobactersp.)、短波单胞菌属(brevundimonassp.)、不粘柄菌属(asticcacaulissp.)、鞘氨醇单胞菌属(sphingomonassp.)、根瘤菌属(rhizobiumsp.)、剑菌属(ensifersp.)、慢生根瘤菌属(bradyrhizobiumsp.)、温单胞菌属(tepidimonassp.)、温胞菌属(tepidicellasp.)、水杆菌属(aquabacteriumsp)、污泥单胞菌属(pelomonassp.)、产碱杆菌属(alcaligenissp.)、无色杆菌属(achromobactersp.)、劳尔氏菌属(ralstoniasp.)、湖沉积杆菌属(limnobactersp.)、马赛菌属(massiliasp.)、氢噬胞菌属(hydrogenophagasp.)、食酸菌属(acidovoraxsp.)、弯曲杆菌属(curvibactersp.)、代尔夫特菌属(delftiasp.)、红育菌属(rhodoferaxsp.)、另类希灭氏菌属(alishewanellasp.)、寡养单胞菌属(stenotrophomonassp.)、独岛菌属(dokdonellasp.)、甲基弯曲菌属(methylosinussp.)、生丝微菌属(hyphomicrobiumsp.)、甲基硫单胞菌属(methylosulfomonassp.)、甲基杆菌属(methylobacteriasp.)、假单胞菌属(pseudomonassp.)如门多萨假单胞菌(pseudomonasmendocina)、肠球菌属(enterococcussp.)、香味菌属(myroidessp.)、伯克霍尔德氏菌属(burkholderiasp.)、产碱菌属(alcaligenessp.)、黄色葡萄球菌属如金黄色葡萄球菌(staphylococcusaureus),埃希氏菌属如大肠杆菌,以及其混合物。根据一种实施方案,该至少一种霉菌菌株选自支顶孢属(acremoniumsp.)、链格孢属(alternariasp.)、曲霉属(aspergillussp.)如黑色曲霉(aspergillusniger)、短柄霉属(aureobasidiumsp.)如出芽短柄霉(aureobasidiumpullulans)、枝孢属(cladosporiumsp.)、镰孢属(fusariumsp.)、毛霉菌(mucorsp.)、青霉属(penicilliumsp.)如绳状青霉(penicilliumfuniculosum)、根霉属(rhizopussp.)、葡萄穗霉属(stachybotryssp.)、木霉属(trichodermasp.)、蛛网菌属(dematiaceaesp.)、茎点霉属(phomasp.)、散囊菌属(eurotiumsp.)、帚霉属(scopulariopsissp.)、短柄霉属(aureobasidiumsp.)、念珠霉属(moniliasp.)、葡萄孢属(botrytissp.)、匍柄霉属(stemphyliumsp.)、壳霉属(chaetomiumsp.)、菌丝体属(myceliasp.)、链孢霉菌属(neurosporasp.)、单格孢属(ulocladiumsp.)、拟青霉属(paecilomycessp.)、节担菌属(wallemiasp.)、弯孢属(curvulariasp.)、以及其混合物。根据一种实施方案,该至少一种酵母菌株选自酵母亚门(saccharomycotina)、外囊菌亚门(taphrinomycotina)、裂殖酵母纲(schizosaccharomycetes)、担子菌门(basidiomycota)、伞菌亚门(agaricomycotina)、银耳纲(tremellomycetes)、柄绣菌亚门(pucciniomycotina)、微球黑粉菌纲(microbotryomycetes)、假丝酵母属(candidasp)如白色假丝酵母(candidaalbicans)、热带假丝酵母(candidatropicalis)、类星形念珠菌(candidastellatoidea)、光滑念珠菌(candidaglabrata)、克鲁斯氏假丝酵母(candidakrusei)、季也蒙氏假丝酵母(candidaguilliermondii)、维斯氏假丝酵母(candidaviswanathii)、葡萄牙假丝酵母(candidalusitaniae)以及其混合物、耶氏酵母属(yarrowiasp.)如解脂耶氏酵母(yarrowialipolytica)、隐球酵母属(cryptococcussp.)如格特隐球菌(cryptococcusgattii)以及新型隐球菌(cryptococcusneofarmans)、接合糖酵母属(zygosaccharomycessp.)、红酵母属(rhodotorulasp.)如胶红酵母(rhodotorulamucilaginosa)、以及其混合物。根据一种实施方案,该至少一种藻类菌株选自地木耳(nostoccommune),蓝藻(gloeocapsaalpicola)(同义字anacystismontana)、软克里藻(klebsormidiumflaccidum)、杆裂丝藻(stichococcusbacillaris)、羊角月芽藻(pseudokirchneriellasubcapitata)、近具刺链带藻(desmodesmussubspicatus)、小皮舟形藻(naviculapelliculosa)、鱼腥藻(anabaenaflosaquae)、聚藻蓝藻(synechococcusleopoliensis),以及其混合物。根据本发明的一种优选实施方案,该至少一种细菌菌株选自大肠杆菌(escherichiacoli)、金黄色葡萄球菌(staphylococcusaureus)、恶臭假单胞菌(pseudomonasputida)、门多萨假单胞菌(pseudomonasmendocina)、食油假单胞菌(pseudomonasoleovorans)、萤光假单胞菌(pseudomonasfluorescens)、产碱假单胞菌(pseudomonasalcaligenes)、类产碱假单胞菌(pseudomonaspseudoalcaligenes)、虫媒假单胞菌(pseudomonasentomophila)、细菌性斑点病菌(pseudomonassyringae)、外链甲基杆菌(methylobacteriumextorquens)、耐辐射甲基杆菌(methylobacteriumradiotolerants)、二氯甲烷甲基杆菌(methylobacteriumdichloromethanicum)、嗜有机甲基杆菌(methylobacteriumorganophilu)、扎维生丝霉菌(hyphomicrobiumzavarzini)、粪肠球菌(enterococcusfaecalis)、香味菌(myroidesodoratus)、绿脓假单胞菌(pseudomonasaeruginosa)、稻皮假单胞菌(pseudomonasorizyhabitans)、洋蒽伯克氏菌(burkholderiacepacia)、粪产碱杆菌(alcaligenesfaecalis)以及少动鞘氨醇单胞菌(sphingomonaspaucimobilis)以及其混合物和/或该至少一种霉菌菌株选自绳状青霉(penicilliumfuniculosum)、黑色曲霉(aspergillusniger)、出芽短柄霉(aureobasidiumpullulans)、互格链格孢(alternariaalternate)、芽枝状枝孢(cladosporiumcladosporioides)、茎点霉菌(phomaviolaceae)、黑细基格孢菌(ulocladiumatrum)、杂色曲霉(aspergillusversicolor)、黑葡萄穗霉(stachybotrischartarum)、产紫青霉(penicilliumpurpurogenum)、胶红酵母(rhodotorulamucilaginosa)和/或该至少一种酵母菌株选自白色假丝酵母(candidaalbicans)和/或该至少一种藻类菌株选自地木耳(nostoccommune),蓝藻(gloeocapsaalpicola)(同义字anacystismontana)、软克里藻(klebsormidiumflaccidum)、杆裂丝藻(stichococcusbacillaris)、羊角月芽藻(pseudokirchneriellasubcapitata)、近具刺链带藻(desmodesmussubspicatus)、小皮舟形藻(naviculapelliculosa)、鱼腥藻(anabaenaflosaquae)、聚藻蓝藻(synechococcusleopoliensis),以及其混合物。本发明的经表面处理的经表面反应的碳酸钙可被引入到制品中以提供具有增强的抗微生物活性的制品。根据本发明的进一步的方面,提供包含通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙或包含其的组合物的制品,其中该制品选自纸产品、工程木材产品、石膏板产品、聚合物产品、卫生用品、医用产品、卫生保健产品、过滤产品、织造材料、非织造材料、土工织物产品、农业产品、园艺产品、衣物、鞋类产品、行李产品、家用产品、工业产品、包装产品、建筑产品、以及构造产品。控制气味的方法根据本发明,提供通过使通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙与有气味的物质接触来控制气味的方法,其中步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为铜盐,选自硝酸铜、硫酸铜、醋酸铜、氯化铜、氟化铜、溴化铜、其水合物及其混合物且最优选选自硫酸铜、其水合物及其混合物。根据本发明的另一种实施方案,提供通过接触可通过包括以下步骤的方法获得的经表面处理的经表面反应的碳酸钙来控制气味的方法:(i)提供至少一种经表面反应的碳酸钙,其中该经表面反应的碳酸钙是天然研磨或沉淀碳酸钙与二氧化碳和一种或多种h3o+离子供体在水性介质中的反应产物,其中该二氧化碳通过h3o+离子供体处理原位形成和/或从外部来源供应,(ii)提供至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物,其中该水溶性金属盐选自铬盐、锰盐、铁盐、钴盐、铜盐、锌盐、银盐及其混合物,(iii)提供水性溶剂,(iv)使步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物与步骤(iii)的该水性溶剂接触以制备含金属的溶液,(v)在一个或多个步骤中,使步骤(i)的该至少一种经表面反应的碳酸钙与步骤(iv)的该含金属的溶液接触以获得混合物,以及(vi)加热从步骤(v)获得的该混合物至20-250℃的温度以形成包含至少一种水不溶性金属化合物的经表面处理的经表面反应的碳酸钙,该至少一种水不溶性金属化合物在该经表面反应的碳酸钙的表面上由步骤(ii)中作为金属盐、金属氢氧化物、金属氧化物或其混合物提供的金属形成,其中步骤(ii)的该至少一种水溶性金属盐、水溶性金属氢氧化物、水溶性金属氧化物或其混合物为铜盐,选自硝酸铜、硫酸铜、醋酸铜、氯化铜、氟化铜、溴化铜、其水合物及其混合物且最优选选自硫酸铜、其水合物及其混合物。根据本发明的“气味”通常被定义为人类或其他动物通过嗅觉感知的通常在非常低的浓度下的一种或多种挥发的化合物。因此,“有气味的物质”是具有嗅味或气味的化合物,即挥发性足以输送至鼻子上部的嗅觉系统中。根据本发明优选待控制的气味是引起令人不愉快感觉的气味即恶臭,但是并不局限于此。这种气味可源自有气味的物质,其优选选自在人类和动物体液和分泌物如月经、血液、血浆、腐脓液;阴道分泌物、粘液、乳汁、尿;粪便;呕吐物和汗液中含有的有气味的物质;源自诸如人或动物组织的腐败物;食品如乳制品、肉和鱼以及水果(如榴莲水果)的有气味的物质。根据本发明的一种优选的实施方案,该有气味的物质选自胺如三乙胺,二乙胺,三甲胺,二氨基丁烷,四亚甲基二胺,五亚甲基二胺,吡啶,吲哚,3-甲基吲哚;羧酸如丙酸,丁酸,3-甲基丁酸,2-甲基丙酸,己酸;硫有机化合物如硫醇如甲硫醇,磷有机化合物如甲膦,二甲膦,其衍生物及其混合物;优选地,该有气味的物质为胺,且最优选地,该有气味的物质为二乙胺。根据本发明的一种示例性的实施方案,该有气味的物质为二乙胺或硫醇如2-丙硫醇。根据本发明的控制气味的方法包括使通过根据本发明的方法可获得的经表面处理的经表面反应的碳酸钙与有气味的物质接触的步骤。该接触可通过本领域技术人员已知的任何方法制备。例如,可使通过根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙与有气味的物质混合。根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙可以固体形式(如作为粉末)或以悬浮液(如水性悬浮液)的形式提供。根据一种优选的实施方案,根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙可以固体形式(如作为粉末)提供。另外可选地,根据本发明的方法可获得的该经表面处理的经表面反应的碳酸钙可被涂覆到可能的材料(如纸或塑料材料)上或被引入到所述材料中,并且之后可使该材料与有气味的物质接触。具体实施方式基于旨在说明本发明的某些实施方案且并非限制性的以下实施例将会更好地理解本发明的范围和益处。实施例1.测量方法在下文中描述了在实施例中实施的测量方法。布氏粘度布氏粘度通过使用配备有合适圆盘锭子(例如锭子2-5)的型号rv的布氏粘度计在25℃±3℃下在100rpm下搅拌1分钟之后来测量。固体含量悬浮液固体含量(也被称作“干重”)通过使用具有以下设置的moistureanalysermj33(瑞士mettler-toledo)测定:150℃的干燥温度,如果质量在30秒的周期内变化不超过1mg,则自动断开,5g至20g悬浮液的标准干燥)。粒子尺寸分布使用malvernmastersizer2000激光衍射系统评价体积中值颗粒直径d50。使用malvernmastersizer2000激光衍射系统测量的d50或d98值指示的直径值使得分别为50%或98%体积的粒子具有小于该值的直径。通过测量获得的原始数据使用米氏理论、以粒子折射率1.57和吸收指数0.005进行分析。重量确定中值粒子尺寸d50(重量)通过沉降法测量,所述沉降法是在重力场中的沉降行为的分析。使用美国micromeriticsinstrumentcorporation的sedigraphtm5100或5120进行测量。方法和仪器为本领域技术人员所知且常见地用于确定填料和颜料的粒子尺寸分布。在0.1%重量na4p2o7水溶液中进行测量。使用高速搅拌器和超声分散样品。比表面积(ssa)通过将样品在真空下于100℃下加热30分钟的时间周期进行调理之后,通过使用来自micromeritics的asap2460仪器使用氮气根据iso9277:2010经由bet方法测量比表面积。在这种测量之前,样品在布氏漏斗内过滤,用去离子水冲洗并且在烘箱中于90-100℃下干燥过夜。随后,将干燥饼在研钵中充分研磨,且将所得粉末在130℃下在湿度平衡下放置,直到达到恒重为止。粒子内侵入式比孔容(以cm3/g表示)使用micromeriticsautoporev9620汞孔率计,使用汞侵入孔隙率测定法测量结果测量比孔容,所述汞孔率计具有最大施加汞压为414mpa(60000psi),等效于0.004μm(~nm)的拉普拉斯喉径。在每个压力步骤使用的平衡时间是20秒。将样品材料密封在3cm3室的粉末透度计中用于分析。使用软件pore-comp(gane,p.a.c.,kettle,j.p.,matthews,g.p.和ridgway,c.j.,“voidspacestructureofcompressiblepolymerspheresandconsolidatedcalciumcarbonatepaper-coatingformulations”,industrialandengineeringchemistryresearch,35(5),1996年,第1753-1764页),针对汞压缩、透度计膨胀和样品材料压缩来校正数据。在累积侵入数据中见到的总孔体积可被分成两个区域,其中从214μm降至约1-4μm的侵入数据显示具有强烈贡献的任何附聚结构之间的样品的粗填充。在这些直径之下的是粒子自身的精细粒子间填充。如果它们也具有粒子内孔,则此区域显现双峰,并且通过获取由汞侵入比峰转折点更细(即比双峰拐点更细)的孔的比孔容,因而定义比粒子内孔体积。这三个区域的总和给出了粉末的总全部孔体积,但强烈地取决于原始样品压实/在分布的粗孔末端处的粉末的沉降。通过获取累积侵入曲线的第一导数,揭示了基于等效拉普拉斯直径的孔尺寸分布,其必然包括孔屏蔽。微分曲线清楚地显示了粗附聚孔结构区域、粒子间孔区域和粒子内孔区域(如果存在的话)。已知粒子内孔直径范围,则可以从总孔体积中减去剩余粒子间和附聚体间孔体积,以给出在每单位质量孔体积(比孔容)方面的单独的内部孔的希望的孔体积。当然,相同的减法原理也适用于分离任何感兴趣的其他孔尺寸区域。水吸取在本文中所指的材料的水分吸取敏感性(moisturepick-upsusceptibility)”在+23℃(±2℃)温度下分别暴露于10%和85%相对湿度的气氛2.5小时之后来确定,以mg水分/g表示。为此目的,样品首先在10%相对湿度的气氛中保持2.5小时,然后将该气氛变为85%相对湿度,在此相对湿度下将样品保持另外2.5小时。在10%和85%相对湿度之间的重量增加被用来计算以mg水分/g样品表示的水分吸取。含湿量在220℃下在氮气下(流量80ml/分钟,加热时间10分钟),在karl-fischercoulometer(c30烘箱:mettlertoledostromboli,mettlertoledo,瑞士)测定含湿量。使用hydranal-waterstandardkf-oven(sigma-adrich,德国)(在220℃测量)检查结果的准确度。x射线衍射(xrd)使用可旋转pmma支撑环在样品上进行xrd实验。使用brukerd8advance粉末衍射仪遵守布拉格定律(bragg'slaw)分析样品。此衍射仪由2.2kwx射线管、样品固持器、θ-θ-测角计和检测器构成。在所有实验中均采用镍过滤的cukα辐射。特征分布(profiles)为在2θ中使用每分钟0.7°的扫描速度自动记录的图表。使用diffracsuite软件包eva和search,基于icddpdf2数据库的参考图,所得粉末衍射图可容易地通过矿物含量分类。衍射数据的定量分析指的是确定多相样品中不同相的量,且已使用diffracsuite软件包topas进行。详细地,定量分析允许使用来自实验数据本身的可量化数字精度来测定结构特性和相比例。这涉及全衍射图的模型化(rietveld方案),使得计算的图复制实验所得。rietveld法需要图案中所感兴趣的所有相的大致晶体结构的知识。然而,整个图案而非少数选定线的使用产生远好于任何单峰强度基方法的准确度和精度。颜料白度r457颜料白度r457根据iso2469:1994(din53145-1:2000和din53146:2000)使用datacolorelrepho(datacolorag,瑞士)在片剂(在omyapress2000上制备,压力=4巴,15秒)上测量。cielab坐标cielabl*、a*、b*坐标根据eniso11664-4使用datacolorelrepho(datacolorag,瑞士)测量并且硫酸钡作为内标。黄色指数cie坐标使用datacolorelrepho(datacolorag,瑞士)测量。黄色指数(=yi)通过下式计算:yi=100*(rx-rz)/ry)。电感耦合等离子体光发射光谱测定法icp-oes为了确定在该经表面反应的碳酸钙的表面上形成的可从该表面脱离的水不溶性金属化合物的量,确定从该经表面处理的经表面反应的碳酸钙的表面浸出到水中的水不溶性金属化合物的量。通过icp-oes分析来自在反应之后制备经表面处理的经表面反应的碳酸钙的每一个过滤步骤的滤液(对于粉末1)或者在1升水(室温,机械搅拌,2小时)中搅拌100g经表面处理的经表面反应的碳酸钙、过滤并通过icp-oes分析滤液(对于粉末6,洗涤程序重复三次),以确定已经溶于水中的水不溶性金属化合物的量。icp-oes在来自perkinelmer的具有旋风喷雾室(twister)(铜线cu224.7、cu324.752、cu327.393)的optima8300dv设备(双向观测)上进行。将粉末样品(1.0g)溶解于hno3(69%,痕量选择)中并且煮沸5分钟。在冷却之后,用水稀释溶液,过滤(0.20mm)并且通过icp-oes分析。滤液样品用hno3(69%,痕量选择)酸化、过滤(0.20mm)并且通过icp-oes分析。抗微生物表面活性测试细菌大肠杆菌(escherichiacoli)dsm1576和金黄色葡萄球菌(staphylococcusaureus)dsm346菌株的新鲜细菌培养物通过在胰蛋白酶大豆琼脂盘(tsa,no.236950,bectondickinsonandcompany,usa)上稀释划线以及在35℃下培养16至20小时而制备。为了测试抗微生物表面活性,使用如上所述而制备的新鲜细菌,遵循日本标准方案jisz28012000。根据经以下修正的日本标准方案jisz28012000进行涂板、计数以及评估。对于所有涂覆的样品来说,培养后从在培养皿中的测试项目释放细菌,使用无菌drigalski抹刀以培养基按摩测试项目,而不是使用兜包袋(stomacherbag)以手按摩。此外,对于涂覆的样品来说,在分析之前测试项目未经70%乙醇消毒。如在日本标准方案jisz28012000中所述,细菌计数以每个测试项目的菌落形成单元(cfu/测试项目)记载,以10cfu/测试项目作为检测极限(lod)。因此,测试项目的抗微生物活性(r)如在日本标准方案jisz28012000中所述进行计算。对其来说,在35℃下培养24小时之后,使用下式:r=log10(a/b),使用测试项目(b)和未处理的对照组(a)上的活细菌平均数来计算抗微生物活性(r)。如果检测到cfu为0,则10cfu/测试项目的值被用于计算抗微生物活性的检测极限。耐真菌生长测试通过以下方式制备新鲜真菌培养物(黑色曲霉(aspergillusniger)(dsm1975)):以孢子和/或真菌菌丝体接种包含1.5%重量琼脂(no.05039,fluka,瑞士)的麦芽琼脂盘(麦芽萃取物培养基,no.1.05397,merckkgaa,德国),并且在25℃下培养直到麦芽琼脂盘完全被孢子覆盖(大约1周)。这种培养技术为本领域技术人员熟知,并且例如被描述于astmd5590-00中。将来自新鲜真菌麦芽琼脂盘的孢子圈接种于麦芽萃取物培养基(no.1.05397,merckkgaa,德国)上。通过混合分散孢子,直到看不到团块为止。将测试项目切成2.5cmx9cm,并且浸于孢子分散体中,排水,并置于50ml具有可透气过滤器的生物反应器(例如tppbioreactors,tpp,瑞士)管中。生物反应器中的测试项目直立地在90%相对湿度和28℃下培养。在不同的培养时间之后,类似于astmd3273-d12评级系统为真菌性污损(defacement)的百分比进行评级。评级10=0污损(未检测到生长)。评级9=1至10%污损。评级8=11至20%污损。评级7=21至30%污损。评级6=31至40%污损。评级5=41至50%污损。评级4=51至60%污损。评级3=61至70%污损。评级2=71至80%污损。评级1=81至90%污损。评级0=91至100%污损。抗藻类效力测试使用杆裂丝藻(stichococcusbacillaris)作为测试生物,根据测试标准dinen15458:2007(油漆和清漆-测试防藻类涂层中膜防腐剂效力的实验室测试方法)测定抗藻类效力。半定量测试方法的原理是将包含膜防腐剂的涂覆样品或未处理的对照组置于营养素琼脂表面(涂覆面朝上)。然后,将标准藻孢子悬浮液接种于表面,并且培养。在四个不同时间点(14、21、28和35天后),评估在琼脂(围绕测试片)上藻类生长和在涂覆样品的表面上藻类生长的强度并且使用以下评级系统与对照组进行比较。0:涂覆样品的表面上没有藻类生长1:包含经改性矿物基填料的涂覆样品相较于包含未处理矿物的样品藻类生长较少。2:包含经改性矿物基填料的涂覆样品相较于包含未处理矿物的样品藻类生长相等或更多。该测试标准在稍微修改的情况下进行三次:1)所有涂覆样品并未根据en23270在23+/-2℃和50+/-5%相对湿度下调理5天,而是在未控制湿度下在23+/-2℃下储存数周。2)所有涂覆样品在测试前未消毒。3)所有涂覆样品的尺寸和形状为矩形(25mmx50mm),而不是圆形(直径55mm)。4)最终评估在第32天进行。5)仅测试针对绿藻杆裂丝藻(stichococcusbacillaris)且并不是针对由标准定义的绿藻和蓝藻的混合物的抗藻类效力。2.含金属的溶液的制备溶液1在玻璃烧杯中提供80g去离子水并且在强烈搅拌下缓慢添加39g硫酸铜(五水合物,sigma-aldrich,西班牙)。将得到的深蓝色混合物在室温下搅拌2小时并且然后过滤。所获得的溶液的固体含量基于该溶液的总重量计为20-24%重量。溶液2硫酸锌的饱和溶液通过在去离子水(215g)中添加七水硫酸锌(350g,sigma-aldrich,美国)来制备。将得到的无色混合物在室温下搅拌2小时并且然后过滤。所得到的溶液的固体含量基于该溶液的总重量计为39%重量。3.经表面处理的经表面反应的碳酸钙的制备除非另外指出,在以下的实施例和对比实施例的制备说明中,“份”形式的重量指示总是指“重量份”。用在以下实施例中的经表面反应的碳酸钙srcc显示以下的特性:d50=2.6μm,bet比表面积=34.7m2/g,以及0.305cm3/g的粒子内侵入式比孔容(对于0.004-0.19μm的孔直径范围来说)。3.1.实施例1–粉末1将处于去离子水(600ml)中的150g经表面反应的碳酸钙(srcc)的悬浮液放置在配备有冷凝器和加料漏斗的1升圆底烧瓶中。将所述混合物加热至回流并且向所述混合物逐滴添加150g之前制备的溶液1(对应于30g硫酸铜的20%重量固体含量的溶液)。所述悬浮液颜色变绿,并且在添加完成后继续再加热2小时(伴随在700rpm下的搅拌下)。然后停止加热并且在布氏漏斗上过滤悬浮液,并且用1升去离子水洗涤。滤液为无色的,并且滤饼(绿色粉末)在1升去离子水中再次分散、搅拌并再次过滤。该洗涤程序重复三次,并且所获得的滤饼然后在烘箱(90℃,减压)中干燥。所获得的绿色粉末(粉末1)然后通过xrd进行分析。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.2克cuso4/克srcc5.8mgcuso4/m2srcc3.2.实施例2–粉末2将200g干燥的经表面反应的碳酸钙(srcc)和5g粉末1放置在高速混合器(mti混合器,mtimischtechnikinternationalgmbh,德国)中,并且混合物通过搅拌10分钟(500rpm,30℃)均质化。在该时间之后,收集所述混合物。获得微绿色的均匀粉末(粉末2)。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.005克cuso4/克srcc0.15mgcuso4/m2srcc3.3.实施例3–粉末3将500g干燥的经表面反应的碳酸钙(srcc)放置在高速混合器(mti混合器,mtimischtechnikinternationalgmbh,德国)中,并且通过搅拌调理10min(3000rpm,120℃)。在该时间之后,引入相对于100份srcc为1份的硫酸铜(具有20%重量固体含量的25g溶液1)并且再继续搅拌20分钟(120℃,3000rpm)。在该时间之后,再引入相对于100份srcc为1附加份的硫酸铜(20%重量固体含量的25g溶液1)并且再继续搅拌20分钟(120℃,3000rpm)。在该时间之后,使混合物冷却并收集。获得微绿色的均匀粉末(粉末3)。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.02克cuso4/克srcc0.58mgcuso4/m2srcc3.4.实施例4–粉末4将处于去离子水(300ml)中的200g经表面反应的碳酸钙(srcc)的悬浮液放置在配备有冷凝器和加料漏斗的1升圆底烧瓶中。向该混合物添加na2co3(0.25mol,100ml去离子水中溶解26.5g)并且将该混合物加热至回流并且向该混合物逐滴添加150ml之前制备的溶液1(0.25mol五水硫酸铜,62.5g五水硫酸铜溶解在200ml去离子水中)。该悬浮液的颜色变绿,并且在添加完成后在800rpm下搅拌再继续加热3小时回流。然后停止加热并且在布氏漏斗上过滤悬浮液,并且用1.5升去离子水洗涤3次。然后所获得的滤饼在1升去离子水中再分散,并且使用以下条件用geaniromobileminor喷雾干燥机进行喷雾干燥:入口温度:200℃出口温度:>85℃喷雾压力:6巴根据出口温度调节泵速。所获得的绿色粉末(粉末4)然后通过xrd进行分析。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.2克cuso4/克srcc5.8mgcuso4/m2srcc3.5.实施例5–粉末5将350g干燥的经表面反应的碳酸钙(srcc)放置在高速混合器(mptischmischer,somakonverfahrenstechnikug,德国)中,并且通过搅拌调理10min(500rpm,120℃,刮刀速度50%,具有用于水分蒸发的敞开出口)。在该时间之后,搅拌速度增加至600rpm,并且在2分钟内向所述混合物添加3.5g的20%重量的溶液1(相当于0.2份/100份srcc)。然后搅拌速度逐渐增加至1000rpm,并且在120℃下继续搅拌20分钟的总时间。在该时间之后,使混合物冷却并收集。获得非常微绿的均匀粉末(粉末5)。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.002克cuso4/克srcc0.058mgcuso4/m2srcc3.6.实施例6–粉末6将300g干燥的经表面反应的碳酸钙(srcc)放置在高速混合器(mptischmischer,somakonverfahrenstechnikug,德国)中,并且通过搅拌调理10分钟(500rpm,120℃,刮刀速度50%,具有用于水分蒸发的敞开出口)。在该时间之后,向所述混合物添加150g的20%重量的溶液1(对应于10份/100份改性碳酸钙)。在120℃下继续搅拌速度20分钟的总时间。在该时间之后,使混合物冷却并收集。获得绿色均匀粉末(粉末6)。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.1克cuso4/克srcc2.9mgcuso4/m2srcc3.7.实施例7–粉末7将处于去离子水(3升)中的400g经表面反应的碳酸钙(srcc)的悬浮液放置在5升烧杯中。将该混合物加热至80℃持续1h(搅拌300rpm)并且向所述混合物逐滴添加194.6g之前制备的溶液3(41.1%重量固体含量的溶液,对应于80g干燥的硝酸铜)。悬浮液颜色变绿,并且在完成添加后再继续加热2小时(伴随在300rpm下搅拌)。然后停止加热并且将悬浮液在布氏漏斗上过滤,并且用3升去离子水洗涤。该滤液是无色的,并且再次将滤饼(绿色粉末)在3升去离子水中再分散,再次搅拌并过滤。所获得的滤饼然后在烘箱中干燥(120℃,7h)。然后通过xrd分析所获得的绿色粉末(粉末7)。铜盐的量(基于无水硫酸铜和处理之前的经表面反应的碳酸钙的计算量计):0.2克cuso4/克srcc5.8mgcuso4/m2srcc3.8.对比实施例1–粉末c1对比实施例1是没有进一步处理的经表面反应的碳酸钙(srcc)(粉末c1)。3.9.对比实施例2–粉末c2将处于去离子水(600ml)中的150g经表面反应的碳酸钙(srcc)的悬浮液放置在配备有冷凝器和加料漏斗的1升圆底烧瓶中。将该混合物加热至回流2小时(伴随在700rpm下搅拌)。然后停止加热并且在布氏漏斗上过滤该悬浮液,并用1升去离子水洗涤。该滤液是无色的,并且然后滤饼(白色粉末)在烘箱中干燥(90℃,减压)。获得白色粉末(粉末c2)。表1:使用铜盐的所制备的经表面处理的经表面反应的碳酸钙的概述表2:粉末c1、1和4的定量rietveld分析(xrd)矿物化学式粉末c1粉末1粉末4方解石caco373.760.364.5羟磷灰石ca5(oh)(po4)326.323.325.1石膏caso4·2h2o6.3-辉铜矿cu2s0.2-水胆矾cu4so4(oh)62.12.7一水蓝铜矾cu4(so4)(oh)6·h2o2.4-钙铜矾cacu4(so4)2(oh)6·3h2o2.7-水合磷酸氢铜cuhpo4·h2o2.7-孔雀石cu2co3(oh)2/7.7其他成分-/-总量100100100数据标准化为100%结晶材料。表3:粉末c1、3、4和5的亮度分析(baso4)样品粉末c1粉末3粉末4粉末5rx(%)97.789.676.596.1ry(%)96.991.882.496.3rz(%)95.791.682.395.2r457tappi(%)95.791.983.095.3黄色指数1.5-1.8-5.61.1cielabl*98.896.892.998.5cielaba*-0.03-3.04-8.99-0.50cielabb*0.810.290.690.77表4:粉末c1和1-5的水吸取样品水吸取(mg/g)c121.4121.92未检出3未检出422.5522.0电感耦合等离子体光发射光谱测定法(icp-oes)如下确定从经表面处理的经表面反应的碳酸钙粉末1和6的表面浸出到水中的铜的量:粉末1:为了确定铜浸出到水中的量,来自反应之后每个过滤步骤(参见粉末1的制备)的滤液通过icp-oes分析以确定已经溶解在水中的铜的量。粉末6:为了确定铜浸出到水中的量,来自本发明的100g粉末在1升水中搅拌(室温,机械搅拌器,2小时)。在该时间之后,将该混合物过滤并分析滤液。该程序重复几次。表4b:来自粉末1和6的洗涤程序的滤液的icp-oes分析。表4c:洗涤步骤之后经过滤的粉末的组成粉末中的cu量(%重量)粉末63.4粉末79.23.10实施例8–粉末8在室温下将420g干燥的经表面反应的碳酸钙(srcc)放置在混合器(m5r-mk,gebrüdermaschinenbaugmbh,德国)中。启动搅拌并且利用蠕动泵逐滴添加(添加时间:约1小时)相对于100份改性碳酸钙来说为4.8份的七水硫酸亚铁(sigmaaldrich,印度,90g之前制备的22.3%重量的水溶液)并且添加后在室温下继续搅拌20分钟。该处理水平对应于约1.38mg/m2。在该时间之后,将该混合物从混合器中取出。收集到米色/棕色粉末(粉末8)。3.11.实施例9–粉末9将处于去离子水(3升)中的400g经表面反应的碳酸钙(srcc)的悬浮液放置在5升烧杯中。将混合物加热至80℃持续1h(搅拌300-400rpm)并且向所述混合物逐滴添加254g饱和七水硫酸亚铁(ii)溶液(23.6%重量固体含量的溶液,对应于相对于100份经表面反应的碳酸钙来说为20份的干燥硫酸亚铁)。悬浮液颜色变为棕色,并且在添加完成后再继续加热2小时(伴随在300-400rpm下的搅拌)。然后停止加热并且在布式漏斗上过滤悬浮液。滤饼在2升去离子水中再分散2次,搅拌并再次过滤。所获得的滤饼然后在烘箱中干燥(120℃,7小时)。然后所获得的棕色粉末(粉末9)通过xrd进行分析。铁盐的量(基于硫酸亚铁和处理之前的经表面反应的碳酸钙的计算量计):0.2克硫酸亚铁/克srcc5.8mg硫酸亚铁/m2srcc表5:利用铁盐的经表面处理的经表面反应的碳酸钙的概述粉末feso4[份/100份srcc]注释c1--c2-悬浮在水中并干燥84.8干加工920湿加工3.12.实施例10–粉末10将处于去离子水(2.5升)中的300g经表面反应的碳酸钙(srcc)的悬浮液放置在配备有机械搅拌器和加料漏斗的5升烧杯中。将混合物加热至80℃并且向该混合物逐滴添加153.8g之前制备的溶液2(39%重量固体含量的溶液,对应于60g硫酸锌)。悬浮液保持白色,并且在添加完成后再继续加热2小时(在400rpm下搅拌)。然后停止加热并且将悬浮液在布式漏斗上过滤。该滤液是无色的,并且再次在2升去离子水中再分散滤饼(白色粉末)、搅拌并再次过滤。该洗涤程序重复第二次,并且所获得的滤饼然后在干燥烘箱中干燥(120℃)。获得白色粉末(粉末10)。锌盐的量(基于无水硫酸锌和处理之前的经表面反应的碳酸钙的计算量计):0.2克znso4/克srcc5.8mgznso4/m2srcc3.13.实施例11–粉末11将处于去离子水(2.5升)中的300g经表面反应的碳酸钙(srcc)悬浮液放置在配备有机械搅拌器和加料漏斗的5升烧杯中。在室温下向该混合物首先添加39.3g碳酸钠(无水,fluka,德国)。然后将混合物加热至80℃并且向该混合物逐滴添加153.8g之前制备的溶液2(39%重量固体含量的溶液,对应于60g硫酸锌)。悬浮液保持白色,并且在添加完成后再继续加热2小时(在400rpm下搅拌)。然后停止加热并且将悬浮液在布式漏斗上过滤。滤液为无色的,并且再次在2升去离子水中再分散滤饼(白色粉末)、搅拌并再次过滤。该洗涤程序重复第二次,并且所获得的滤饼然后在干燥烘箱中干燥(120℃)。获得白色粉末(粉末11)。锌盐的量(基于无水硫酸锌和处理之前的经表面反应的碳酸钙的计算量计):0.2克znso4/克srcc5.8mgznso4/m2srcc3.14.实施例12–粉末12将300g干燥的经表面反应的碳酸钙(srcc)放置在高速混合器(mptischmischer,somakonverfahrenstechnikug,德国)中,并且通过搅拌调理10分钟(600rpm,120℃,刮刀速度50%,具有用于水分蒸发的敞开出口)。在该时间之后,向所述混合物逐滴添加76.9g之前制备的硫酸锌溶液(39%重量固体含量的溶液,对应于30g硫酸锌)。搅拌速度在120℃下继续20分钟的总时间。在该时间之后,使混合物冷却并收集。获得白色均匀粉末(粉末12)。锌盐的量(基于无水硫酸锌和处理之前的经表面反应的碳酸钙的计算量计):0.1克znso4/克srcc2.9mgznso4/m2srcc表6:制备的经表面处理的经表面反应的碳酸钙的概述粉末znso4[份/100份srcc]na2co3[份/100份srcc]注释c1---c2--悬浮在水中并干燥1020-湿加工112013湿加工1210-干加工4.气味控制:4.1仪器:-吸着管(sigmaaldrich,不锈钢,1/4英寸×31/2英寸)-具有tenaxta的热解吸管(sigmaaldrich,不锈钢,1/4英寸×31/2英寸)-袖珍泵210-1000系列(skcinc,eightyfour,pa,usa)-td-gc-ms(热解吸气相色谱质谱仪):tdturbomatrix650perkinelmer温度模式:管250℃,阀250℃,传输200℃,捕集阱低温-20℃,捕集阱高温300℃定时:解吸10min,吹扫1.0min,捕集阱维持5.0min气动设定:柱160kpa,出口分流40ml/min,进口分流20ml/min,解吸30ml/mingc方法clarus680perkinelmer柱:optima5,accent1.0μm,60m*0.32mm,macherey-nagel炉温:110℃持续10minmsclarussq8tperkinelmer溶剂延迟0.0min。用于二乙胺分析的全扫描25-350m/z(质量/电荷)(ei+)用于2-丙硫醇分析的sir扫描61,76(ei+)4.2有气味的物质:二乙胺(sigmaaldrich,cas109-89-7)2-丙硫醇(sigmaaldrich,cas75-33-2)4.3二乙胺的有气味的物质的控制:制备6.118g/l二乙胺在水中的储备溶液。为了进行吸收/吸附试验,吸收/吸附管填充有以下材料:表7:吸收/吸附管的填料材料实施例填料材料o1玻璃棉o260mg的玻璃棉和粉末c1的1:4混合物o360mg的玻璃棉和粉末1的1:4混合物在吸着管之前安装填充有10μl制备的二乙胺基储备溶液的小瓶,在该管之后安装具有tenaxta的热解吸管。利用水浴将小瓶在40℃下加热1分钟并且然后在室温下(23℃)在80ml/min的速率下借助于袖珍泵(skc)在5分钟期间通过两个管从有气味的物质填充的小瓶吸取空气。随后,在tenaxta管中的有气味的物质的含量借助于td-gc-ms来分析。检测峰下的面积成比例地对应于有气味的物质(二乙胺)的浓度。因此,被不同材料吸收/吸附或与不同材料反应的有气味的物质可借助于峰面积来比较。试验重复几次。所获得的平均值总结于图1中,该图反映了所得到的各个样品移除有气味的物质的相对效率,其中100%是指针对空白样品确定的最大值(实施例o1)。如从这些结果可清楚地看出的,与未处理的粉末(参见实施例o2)相比,从经表面处理的经表面反应的碳酸钙来移除二乙胺具有改善的效率(参见实施例o3)。4.42-丙硫醇的有气味的物质的控制制备0.200g/l的2-丙硫醇在水中的储备溶液。为了进行吸收/吸附试验,吸收/吸附管填充有以下材料:表8:吸收/吸附管的填料材料实施例填料材料o4玻璃棉o5120mg的玻璃棉和粉末c1的1:4混合物o6120mg的玻璃棉和粉末1的1:4混合物在吸着管之前安装填充有10μl制备的2-丙硫醇基储备溶液的小瓶,在该管之后安装具有tenaxta的热解吸管。利用水浴将小瓶在40℃下加热1分钟,并且然后在室温下(23℃)在80ml/min的速率下借助于袖珍泵(skc)在5分钟期间通过两个管从有气味的物质填充的小瓶吸取空气。随后,在tenaxta管中的有气味的物质的含量借助于td-gc-ms来分析。检测峰下的面积成比例地对应于有气味的物质(2-丙硫醇)的浓度。因此,被不同材料吸收/吸附或与不同材料反应的有气味的物质可借助于峰面积来比较。试验重复几次。所获得的平均值总结于图2中,该图反映了所得到的各个样品移除有气味的物质的相对效率,其中100%是指针对空白样品确定的最大值(实施例o4)。如从这些结果可清楚看出的,含有未处理的经表面反应的碳酸钙(粉末c1)的管显示管之后2-丙硫醇的量没有减少(实施例o5),然而当在管中使用经表面处理的经表面反应的碳酸钙粉末时(实施例o6),在管之后几乎没有2-丙硫醇可被检测到。5.经表面处理的经表面反应的碳酸钙的浆料和纸涂层实施例13-17–浆料13-17以及对比实施例3和4(c3和c4)通过在室温下以930rpm搅拌如下表9中所示组成的混合物10分钟在pendraulik搅拌器上制备浆料。表9:制备的浆料的概述da=分散剂(100%钠中和的聚丙烯酸,mw=3500g/mol,ph=8)。实施例18-22–涂层色料制剂18-22和对比实施例5和6(c5和c6)然后用根据实施例13-17以及对比实施例c3和c4的浆料制备含有100份各个粉末(重量/重量)和6份(干燥/干燥)styronald628(basf,德国)的涂层色料并且涂覆在来自瑞士fischerpapierag的super箔(厚度80μm,尺寸:18×26cm,62g/m2,聚丙烯)上。涂层色料的组成和涂层重量总结于下表10中。表10:涂层色料制剂以及涂层重量6.抗微生物活性实施例23–含有经表面处理的经表面反应的碳酸钙的纸涂层的耐真菌生长测试包含涂层的选定纸样品的抗真菌活性按照在以上测量方法部分“耐真菌生长测试”中所描述的进行测试,所述涂层包含本发明的经表面处理的经表面反应的碳酸钙,根据实施例18-22制备,以及根据c6制备。表11a:以一式三份进行的在28℃和90%相对湿度下培养3天之后在耐真菌生长测试中不同的表面涂布的纸样品由于黑色曲霉(aspergillusniger)(dsc1957)的真菌性污损。1根据astmd3273-d12,三份的平均值表11b1:以一式三份进行的在28℃和90%相对湿度下培养7天之后在耐真菌生长测试中不同的表面涂布的纸样品由于黑色曲霉(aspergillusniger)(dsm1957)的真菌性污损1根据astmd3273-d12,三份的平均值表11b2:以一式三份进行的在28℃和90%相对湿度下培养10天之后在耐真菌生长测试中不同的表面涂布的纸样品由于黑色曲霉(aspergillusniger)(dsm1957)的真菌性污损1根据astmd3273-d12,三份的平均值表11c:以一式三份进行的在28℃和90%相对湿度下培养13天之后在耐真菌生长测试中不同的表面涂布的纸样品由于黑色曲霉(aspergillusniger)(dsm1957)的真菌性污损1根据astmd3273-d12,三份的平均值表11d:以一式三份进行的在28℃和90%相对湿度下培养18、24、28和31天之后在耐真菌生长测试中不同的表面涂布的纸样品由于黑色曲霉(aspergillusniger)(dsm1957)的真菌性污损。对于所有四个时间点来说结果保持不变,但只显示一次。1根据astmd3273-d12,三份的平均值实施例24–纸涂层的抗微生物表面活性包含涂层的选定纸样品的抗微生物活性按照在以上测量方法部分“抗微生物表面活性测试”中所描述的进行测试,所述涂层包含本发明的经表面处理的经表面反应的碳酸钙,根据实施例18-22制备,以及根据对比实施例c5和c6制备。表12-14显示涂布的纸样品18-22以及对比样品c5和c6(未经处理的测试项目和掺合物)的cfu计数/测试项目以及对金黄色葡萄球菌(s.aureus)(表12和13)和大肠杆菌(表14)的计算的抗微生物活性。表12-14中的术语lod指的是检测极限。表12:经表面处理的经表面反应的碳酸钙对金黄色葡萄球菌(s.aureus)的抗微生物活性。n/a:不适用表13:经表面处理的经表面反应的碳酸钙对金黄色葡萄球菌(s.aureus)的抗微生物活性。n/a:不适用表14:经表面处理的经表面反应的碳酸钙对大肠杆菌的抗微生物活性。n/a:不适用实施例25–经表面处理的经表面反应的碳酸钙的抗藻类活性根据实施例18和22制备的含有本发明的经表面处理的经表面反应的碳酸钙的各种类型的涂层的抗藻类效力根据上述测试标准确定。表15显示了测试结果。表15:经表面处理的经表面反应的碳酸钙对绿藻杆裂丝藻(stichococcusbacillaris)的抗微生物活性。根据定义,未处理的测试项的评级为2。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1