一种安全简单制备双掺杂氮和磷碳量子点的方法与流程
本发明涉及碳量子点,特别是涉及一种安全简单制备双掺杂氮和磷碳量子点的方法,属于纳米材料科学领域。
背景技术
碳量子点亦称碳点,是指粒径一般在10nm以内的准球形的碳纳米颗粒,在2004年由xu在电弧放电制备单壁碳纳米管时意外制备出来(j.am.chem.soc.2004,126,12736–12737),并在2006年被sun发现其具有高荧光量子效率(j.am.chem.soc.2006,128,7756–7757),由此逐渐获得人们的关注。与传统有机染料或者量子点相比,碳点对细胞毒性低,生物相容性好,制备方法简单,物理、光学性能好,在生物成像,光催化,生物医药等领域都获得了极大的关注,如作为药物载体,基因运输载体,或荧光探针诊断人体肿瘤组织等都有大量的研究。
制备碳点的方法包括自上而下法(电化学氧化法、电弧法、激光消融法等)和自下而上法(微波辅助法、水热法、强酸氧化等)。但是以上方法或者操作条件复杂苛刻,需要用到特定的设备,或者需要强酸强碱、高温高压条件,具有一定危险性。
目前制备出的碳点缺陷明显,碳点的荧光量子效率较低,发射光也集中在蓝绿光,一定程度限制了碳点的应用。有大量的研究发现可以通过在碳点上掺杂别的原子来使发射峰红移并且提高荧光量子效率,其中研究和制备最多的是掺氮以及磷的碳点。以往研究中大都通过混合碳前驱体和有机胺如乙二胺、聚乙烯亚胺等,在碳点表面引入-nh2来改善荧光性能,然而乙二胺易燃,腐蚀性强,对人体伤害大,聚乙烯亚胺更是有强毒性,应用于生物体内有一定风险,需要寻找另外更安全的材料改善性能。
技术实现要素:
为解决现有碳量子点制备繁琐、荧光性能差,以及掺氮剂毒性较大的问题,本发明提供一种以氨基酸为掺氮源,磷酸为氧化剂,通过超声预处理原料,然后油浴加热制备出氮、磷共掺杂碳点的方法。该制备方法安全简单,绿色环保,碳点的荧光性能好,并且碳点的荧光能够随着氨基酸种类变化发生从蓝光到红光的红移。
本发明氨基酸作为一种生物小分子,安全无毒,来源广泛,更重要的是氨基酸结构存在氨基和羧基,很容易发生缩聚反应,可用于制备碳点,在碳点中掺入氮,并加入磷酸氧化,从而制备出氮、磷共掺杂碳点。
本发明油浴加热具有操作简单,容易控制反应条件等优点,然而目前所见关于油浴法制备碳点的研究很少,特别是在较低温度下油浴加热制备碳点,如能够成功实现,不仅操作更加安全,也能够降低制备碳点的能耗和成本。
一种安全简单制备双掺杂氮和磷碳量子点的方法,包括如下步骤:
1)将氨基酸和碳前驱体溶解在去离子水中,然后再加入磷酸溶液中,搅拌配成澄清溶液;所述的碳前驱体为葡萄糖、一水合柠檬酸或环糊精中的一种或两种;所述的氨基酸和碳前驱体摩尔比为1:2~2:1;
2)将溶液转移至超声波机中进行超声处理,超声时间为1~2h;
3)将超声过后的溶液在80~150℃条件下油浴加热、回流1~5h,溶液由澄清透明逐渐变得棕黄、浑浊,自然冷却到室温,制备出同时双掺杂氮、磷的碳点;
4)对步骤3)冷却后的溶液进行离心,除去大颗粒杂质,清液再在透析袋中透析,除掉未反应的原料和其它小颗粒杂质,得到高纯荧光碳点。
为进一步实现本发明目的,优选地,所述的氨基酸是指人体合成蛋白质所需的20种氨基酸。该氨基酸优选为谷氨酸,天冬氨酸,色氨酸,酪氨酸,丝氨酸和组氨酸中一种;构型为l型。
优选地,所述的磷酸溶液的质量分数80-90%;所述的磷酸溶液和去离子水的体积比为1:1-2.0。
优选地,所述的超声波频率为40-80khz。
优选地,所述的油浴加热时间在1~5h。
优选地,所述的离心的转速范围是1-3万转/分钟。
优选地,所述的透析袋的截留分子量为500-1000da。
相对于现有技术,本发明具有如下优点:
1)以氨基酸替代聚乙烯亚胺,乙二胺等胺类作为掺氮源,来料广泛,安全无毒,并通过加入磷酸氧化制备出氮、磷共掺杂碳点,增强碳点的荧光性能;
2)使用超声波处理联合油浴加热法替代水热法和微波辅助法,避免了高温高压的反应条件,油浴加热可以在低于100℃条件下进行,反应更加安全,并且能够快速、大规模制备碳点;
3)根据氨基酸种类以及氨基酸/碳前驱体的摩尔比、反应时间等的不同,可以获得不同发射不同荧光的碳点,能够满足多种用途的需要;
4)碳点表明具有丰富的羟基,在水中溶解性好,且含有氨基,能够通过化学反应或者氢键作用连接别的物质,应用于细胞检测、人体成像等领域。
附图说明
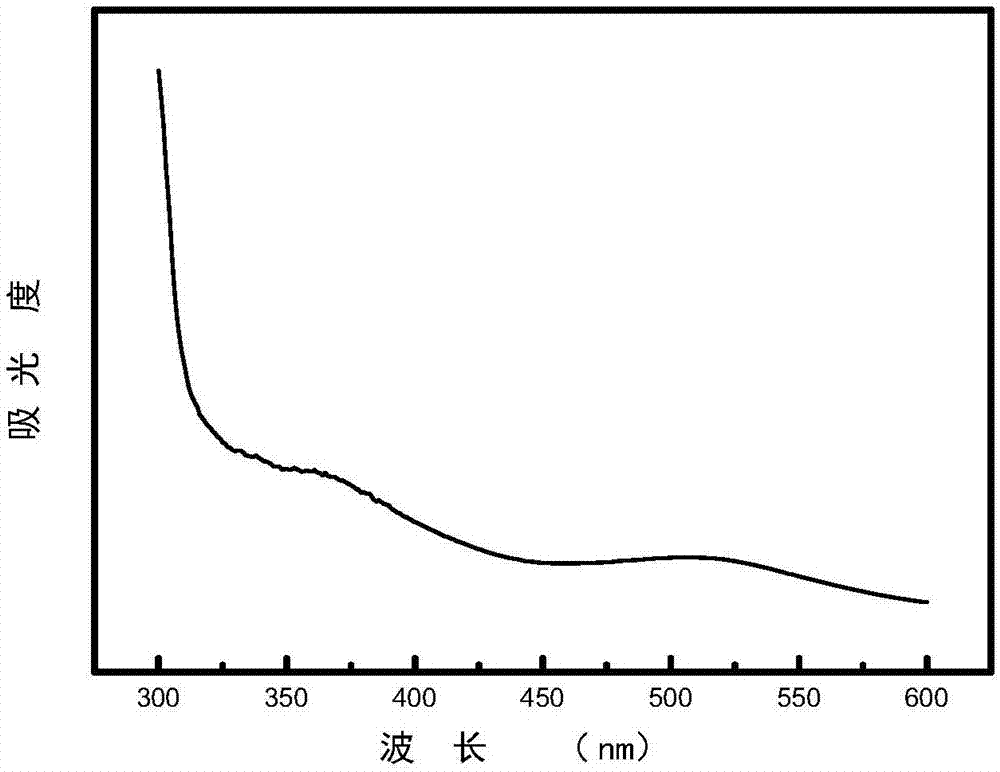
图1为实施例1制备出的色氨酸-碳点的紫外吸收光谱图。
图2为实施例1制备出的色氨酸-碳点的红外吸收光谱图。
图3为实施例1制备出的色氨酸-碳点在510nm激发时的荧光光谱图。
图4为实施例1在510nm激发光激发时不同浓度色氨酸-碳点的荧光光谱。
图5为实施例4在510nm激发光激发时不同氨基酸/葡萄糖摩尔比制备的色氨酸-碳点的荧光光谱。
图6为实施例5在不同加热时间下合成的色氨酸-碳点的荧光光谱图。
图7为实施例6在不同超声时间下合成的色氨酸-碳点的荧光光谱图。
图8为实施例7在不同ph时色氨酸-碳点溶液的荧光光谱图。
图9为实施例8在不同叶酸、色氨酸-碳点比例下溶液的荧光光谱图。
具体实施方式
为更好地理解本发明,下面结合附图和实施例来进一步描述本发明的制备方法及其效果,但本发明的实施方式不限如此。
实施例1
色氨酸-碳点的制备:
称量0.005mol色氨酸和0.005mol葡萄糖倒入烧杯中,加入5ml去离子水,加入5ml质量分数80%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
将烧杯置于超声波机中超声处理2h,超声波频率为40hz。超声后溶液外观上没有变化。
将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为90℃,加热5小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变红,最终变为紫红色。加热完毕后,冷却到室温。碳点经过适当稀释,在太阳光下为红色,在365nm紫外光照射下发橙红荧光。
在10000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为1000da的透析袋中透析,每24h换一次水,共透析两天,去除未反应的原料以及小颗粒的杂质。
图1是色氨酸-碳点的紫外吸收光谱图,表征碳点的紫外吸收特性。碳点在510nm有吸收峰,说明最大吸收波长为510nm。
图2是色氨酸-碳点的红外吸收光谱图,3464cm-1是o-h的伸缩振动,该处的峰强度很大,说明碳点上有较多的羟基,这与碳点在水中的溶解性较好相符合。2375cm-1是p-h的伸缩振动,说明在原料中掺入磷原子,可以在碳点中引入p-h基团,成功得将磷原子掺入到碳点中,提高了碳点的荧光性能。1640cm-1是c=n的伸缩振动,1156cm-1是c-n的伸缩振动,说明氨基酸上的氮原子掺入到碳点上,综上,成功制备出了氮、磷共掺杂的碳点。
图3是在510nm激发时色氨酸-碳点的荧光光谱图,可以看到碳点的发射峰在610nm左右,发出明显的橙红色荧光。
图4是510nm激发时不同浓度色氨酸-碳点的荧光光谱图,可以看出随碳点浓度的增大,其荧光强度下降,发射峰发生红移。
用透射电子显微镜对其形貌和粒径进行表征,可以证明碳量子点是粒径在10nm以内的准球形的碳纳米颗粒。
实施例2
谷氨酸-碳点的制备:
称量0.0005mol谷氨酸和0.005mol葡萄糖倒入烧杯中,加入5ml去离子水,再倒入5ml质量分数85%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
将烧杯置于超声波机中超声处理2h,超声波频率为50khz。
将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为150℃,加热1小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变成棕色。加热完毕后,冷却到室温。获得的高浓度碳点经过稀释后,在太阳光下为浅黄色,在365nm紫外光照射下发黄光。
在20000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为500da的透析袋中透析,每24h换一次水,共透析4天,以去除未反应的原料以及小颗粒的杂质。
实施例3
天冬氨酸-碳点的制备:
称量0.005mol天冬氨酸和0.005mol一水合柠檬酸倒入烧杯中,加入5ml去离子水,加入5ml质量分数90%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
将烧杯置于超声波机中超声处理2h,超声波频率为60khz。超声后溶液外观上没有变化。
将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为120℃,加热2小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变红,最终变为黑棕色。加热完毕后,冷却到室温。碳点经过适当稀释,在太阳光下为棕色,在365nm紫外光照射下发蓝光。
在30000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为800da的透析袋中透析,每24h换一次水,共透析两天,去除未反应的原料以及小颗粒的杂质。
实施例4
不同氨基酸/葡萄糖摩尔比的色氨酸-碳点的制备:
在葡萄糖和色氨酸总浓度为1mol/l条件下,称量葡萄糖和氨基酸摩尔比为2:1,3:2,1:1,2:3,2:1的原料分别置于5个烧杯中,加入5ml去离子水和5ml质量分数85%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
分别将烧杯置于超声波机中超声处理2h,超声波频率为40khz。超声后溶液外观上没有变化。
分别将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为100℃,加热2小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变红,最终变为紫红色。加热完毕后,冷却到室温。碳点经过适当稀释,在太阳光下为红色,在365nm紫外光照射下发橙红荧光。
在10000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为1000da的透析袋中透析,每24h换一次水,共透析两天,去除未反应的原料以及小颗粒的杂质。
图5是510nm激发时不同氨基酸/葡萄糖摩尔比制备的色氨酸-碳点的荧光光谱图,可以看出随着葡萄糖含量的增多,发射峰发生红移,但荧光强度降低随之降低。
实施例5
不同加热时间色氨酸-碳点的制备
称量0.005mol色氨酸和0.005mol葡萄糖倒入烧杯中,加入5ml去离子水,加入5ml质量分数85%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
将烧杯置于超声波机中超声处理2h,超声波频率为40khz。超声后溶液外观上没有变化。
将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为90℃,加热1-5小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变红,最终变为紫红色。加热完毕后,冷却到室温。碳点经过适当稀释,在太阳光下为棕色,在365nm紫外光照射下碳点随加热时间增长,发光由绿光红移至橙光后又蓝移至绿光。
在10000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为1000da的透析袋中透析,每24h换一次水,共透析两天,去除未反应的原料以及小颗粒的杂质。
图6是不同加热时间下合成的碳点的荧光光谱图。可以看出随着加热时间的增加,荧光发射峰强度先增大后减小,加热时间为3h碳点荧光强度达到最大值。
实施例6
不同超声时间色氨酸-碳点的制备:
称量0.005mol色氨酸和0.005mol葡萄糖倒入烧杯中,加入5ml去离子水,加入5ml质量分数85%的磷酸溶液,搅拌至完全溶解,形成澄清透明的溶液;
将烧杯置于超声波机中超声处理40~140min,超声波频率为40khz。超声后溶液外观上没有变化。
将溶液转移到25ml圆底烧瓶中,在恒温加热磁力搅拌器中加热回流,温度保持为90℃,加热2小时,加热过程中,溶液颜色逐渐从澄清透明逐渐变红,最终变为紫红色。加热完毕后,冷却到室温。碳点经过适当稀释,在太阳光下为棕色,在365nm紫外光照射下碳点随加热时间增长,发光由绿光红移至橙光后又蓝移至绿光。
在10000rmp条件下离心10min去除大颗粒杂质,然后再在截留分子量为1000da的透析袋中透析,每24h换一次水,共透析两天,去除未反应的原料以及小颗粒的杂质。
图7是不同加热时间下合成的碳点的荧光光谱图。可以看出随着超声时间的增加,碳点溶液的激发波长和荧光强度基本上呈先增加后减少的趋势,当超声时间为120min时,荧光强度最大。
实施例7
不同ph色氨酸-碳点溶液的配制:
称量实施例1中制备的色氨酸-碳点0.3g入烧杯,称量4份,标号为1、2、3、4,分别加入30mlph=12的naoh溶液、去离子水、ph=4的hcl溶液、ph=2的hcl溶液,搅拌溶解。
图8是不同ph色氨酸-碳点溶液的荧光光谱图。可以看出碳点溶液在碱性条件下荧光强度最低,ph=4时荧光强度最大,说明碳点溶液在弱碱性条件下荧光最强。
实施例8
叶酸-色氨酸-碳点的制备:
称量实施例1中制备的色氨酸-碳点0.3g入烧杯,称量3份,标号为1、2、3,分别加入30ml去离子水。向2号溶液中加入0.05g叶酸,搅拌均匀,3号溶液中加入0.1g叶酸搅拌均匀。
图9是不同叶酸、碳点比例下的溶液的荧光光谱图。可以看出溶液的荧光随着叶酸浓度增加而逐渐减弱,当叶酸和色氨酸-碳点比例上升至1:1时,发生荧光猝灭。说明本技术制得的色氨酸-碳点能够通过化学反应或者氢键作用连接叶酸,使之发生荧光猝灭,因此可应用于细胞检测、人体成像等领域。
上述实施例可见,本发明碳点表明具有丰富的羟基,在水中溶解性好,且含有氨基,能够通过化学反应或者氢键作用连接其他物质例如叶酸,应用于细胞检测、人体成像等领域。与一般碳点相比,本发明碳点具有随浓度的增大,发射峰红移的情况,因此可调节碳点浓度,得到发光为由蓝色到红色的溶液,可应用与多色成像领域。
现有技术制备碳点大多需要强酸强碱、高温高压的条件,具有一定危险性。本发明通过对原料超声预处理然后在低温下进行油浴加热即可制备出碳点,操作简单、安全,能耗低,且能够快速制备出大量碳点。本发明以葡萄糖为前驱体,氨基酸为氮源,磷酸为氧化剂,水为溶剂,原材料来源广泛,且安全无毒,制备出的碳点同时掺氮、磷,碳点的荧光性能好。
本发明根据氨基酸种类以及氨基酸/碳前驱体的摩尔比、反应时间等的不同,可以获得不同发射不同荧光的碳点,能够满足多种用途的需要。
本发明不受上述实施例约束,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的替代方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!