一类自闪烁罗丹明螺硫酯荧光染料及其合成方法和在超分辨成像领域的应用
本发明属于荧光染料领域,具体涉及一类自闪烁罗丹明螺硫酯荧光染料在超分辨成像领域的应用。
背景技术:
2014年诺贝尔化学奖颁给了三个物理学家:ericbetzig、stefanw.hell和w.e.moerner,以表彰他们对于发展超分辨荧光成像技术做出的卓越贡献。超分辨光学显微镜将分辨率极限从几百纳米提升到了几纳米。相比于电子显微镜和扫描电子显微镜,超分辨光学以其独特的非侵入性和高度特异性地多色标记,非常适合于活细胞成像,从而使科学家们能够观察到细胞中不同分子在纳米尺度上的运动。
因此,近年来,超分辨荧光成像技术在生物医学领域得到了迅猛发展。相应地,人们对该项技术所依附的灵魂工具---荧光染料的性能也提出了更高的要求。因为,在进行活细胞超分辨成像时,往往需要用到高强度的激光对染料进行激发,在某些时候还需要加入如硫醇等稳定剂以保证成像质量。而以上因素除了会对细胞的生理活性产生一定的影响外,还要求染料的光稳定性和亮度更高,而大部分传统荧光染料无法满足上述要求。
技术实现要素:
本发明目的之一是提供一类自闪烁罗丹明螺硫酯荧光染料,并应用于活细胞超分辨成像。本发明中的罗丹明螺硫酯荧光染料在细胞内具有无需外加稳定剂、不需要强激活光即可自闪烁的优异性能,适合在活细胞内进行随机光学重构显微成像(storm)应用。
本发明的另一目的是提供一类自闪烁罗丹明螺硫酯荧光染料的制备方法,在罗丹明螺硫酯荧光母体的一端通过哌嗪链接halo蛋白标签的底物分子氯代烷基衍生物,该方法具有步骤简单、容易分离、原料价廉等优点。
该类自闪烁罗丹明螺硫酯荧光染料具有如下结构:
其中:
r1和r2是相同或不同的基团,为h、cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、cmh2m+1co、cmh2m+1so2、cmh2m+1phso2中的任一基团;在上述的不同结构通式的化合物或基团中,m是1~20之间的任何整数;n是0~20之间的整数。
此类自闪烁罗丹明螺硫酯荧光染料的合成路线如下所示:
具体合成步骤如下:
(1)中间体n,n-二取代酮酸的合成:
将3-羟基-r1,r2-二乙基苯胺和邻苯二甲酸酐置于两口烧瓶中,抽真空并用氮气置换三次;在氮气保护下,用注射器向烧瓶中加入甲苯,升温至加热回流8-16h;冷却至室温后,将反应液倒入水中,抽滤,用少量甲醇洗涤滤饼,得到淡粉色固体将得到的固体用异丙醇进行重结晶,重结晶析出的固体用少量甲醇洗涤,得到淡黄色固体即为化合物1的粗产物,即为中间体n,n-二取代酮酸;
(2)中间体单哌嗪取代罗丹明的合成:
将步骤(1)得到的中间体n,n-二取代酮酸和间羟基苯基哌嗪置于两口烧瓶中,抽真空并用氮气置换3次;在氮气保护下用向烧瓶中注入甲磺酸;在90℃下反应过夜;在冰水浴和搅拌条件下,缓慢将冷却后的反应液加入碳酸钠水溶液中,用ph试纸监测调节溶液呈弱碱性;用二氯甲烷对反应混合液进行萃取,合并后的有机相用无水硫酸钠进行干燥。通过减压蒸馏除去溶剂后,剩余物用硅胶柱层析进行提纯,得到红色固体即为化合物2,即为中间体单哌嗪取代罗丹明;
(3)中间体fmoc保护的单哌嗪取代罗丹明的合成:
将步骤(2)得到的中间体单哌嗪取代罗丹明、芴甲氧羰酰琥珀酰亚胺和无水碳酸钠置于两口烧瓶中,抽真空并用氮气置换3次;在氮气保护下用注入乙腈,在室温下反应6-10h;通过减压蒸馏除去溶剂后,用乙酸乙酯将粗产物溶解,抽滤,取滤液;用少量水洗涤滤液,有机相用无水硫酸钠进行干燥;除去溶剂后,剩余物用硅胶柱层析进行提纯,得到粉色固体即为化合物3,即为中间体fmoc保护的单哌嗪取代罗丹明;
(4)中间体fmoc保护的单哌嗪取代罗丹明螺硫酯的合成:
将将步骤(3)得到的中间体fmoc保护的单哌嗪取代罗丹明置于圆底烧瓶中,加入1,2-二氯乙烷使反应物完全溶解;在搅拌条件下向溶液中滴加三氯氧磷后,升温至回流持续反应6-8h;通过减压蒸馏除去溶剂后,剩余物用四氢呋喃溶解,并将其缓慢滴加进含有硫脲、四氢呋喃、三乙胺和水的混合溶液中;在室温下搅拌12-20h后除去溶剂,将剩余物加入水中,用乙酸乙酯对水相萃取,合并后的有机相用无水硫酸钠干燥,之后除去溶剂,粗产物用硅胶柱层析进行提纯,得到白色固体即为化合物4,即为中间体fmoc保护的单哌嗪取代罗丹明螺硫酯;
(5)中间体单哌嗪取代罗丹明螺硫酯的合成:
将步骤(4)得到的中间体fmoc保护的单哌嗪取代罗丹明螺硫酯置于圆底烧瓶中,向烧瓶中加入二乙胺的乙腈溶液,在室温下搅拌5-10h;通过减压蒸馏除去溶剂,剩余物用硅胶柱层析进行提纯,得到紫色固体即为化合物5,即为中间体单哌嗪取代罗丹明螺硫酯;
(6)中间体羧酸烷基取代罗丹明螺硫酯的合成:
将步骤(5)得到的中间体单哌嗪取代罗丹明螺硫酯、末端卤素取代的饱和脂肪羧酸、无水碳酸钾和碘化钾置于圆底烧瓶中,加入适量乙腈使固体溶解,加热回流过夜;通过减压蒸馏除去溶剂,剩余物用硅胶柱层析进行提纯,得到紫色固体即为化合物6,即为中间体羧酸烷基取代罗丹明螺硫酯;
(7)自闪烁罗丹明螺硫酯荧光染料的合成:
将步骤(6)得到的中间体羧酸烷基取代罗丹明螺硫酯、1-羟基苯并三唑(hobt)和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc-hcl)置于圆底烧瓶中,加入超干n,n-二甲基甲酰胺,在搅拌条件下加入halo-nh2和三乙胺,在室温下搅拌2天;通过减压蒸馏除去溶剂后,剩余物用硅胶柱层析进行提纯,得到无色粘稠液体即为自闪烁罗丹明螺硫酯荧光染料。
步骤(1)中:3-羟基-r1,r2-二乙基苯胺和邻苯二甲酸酐的摩尔比为1:0.8-1.2;3-羟基-r1,r2-二乙基苯胺与甲苯的质量体积比为1:5-10(mg:ml)。
步骤(2)中:中间体n,n-二取代酮酸和间羟基苯基哌嗪的摩尔比为1:1.2-2.0;中间体n,n-二取代酮酸与甲磺酸的质量体积比为1:2.0-5.0;碳酸钠水溶液调节反应液的ph值在7.0-8.0之间。
步骤(3)中:中间体单哌嗪取代罗丹明、芴甲氧羰酰琥珀酰亚胺和无水碳酸钠的摩尔比为1:1:1.2-5.0。
步骤(4)中:中间体fmoc保护的单哌嗪取代罗丹明和三氯氧磷的摩尔比为1:3.0-5.0;中间体fmoc保护的单哌嗪取代罗丹明和二氯甲烷的质量体积比为1:5-10(mg:ml);中间体3和硫脲的摩尔比为1:1.2-2.0;中间体fmoc保护的单哌嗪取代罗丹明和三乙胺的摩尔比为1.0:3.0-10.0。
步骤(5)中:中间体fmoc保护的单哌嗪取代罗丹明螺硫酯与15%二乙胺乙腈溶液的质量体积比为1.0:5.0-10.0(mg:ml)。
步骤(6)中:中间体单哌嗪取代罗丹明螺硫酯、末端卤素取代的饱和脂肪羧酸、无水碳酸钾和碘化钾的摩尔比为1.0:1.0-1.2:1.3-1.5:1.5-2.0;中间体单哌嗪取代罗丹明螺硫酯与乙腈的质量体积比为1.0:5.0-10.0(mg:ml)。
步骤(7)中:中间体羧酸烷基取代罗丹明螺硫酯、1-羟基苯并三唑(hobt)、三乙胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc-hcl)和halo-nh2的摩尔比1.0:3.0-5.0:5.0-10.0:3.0-5.0:2.0-3.0;中间体羧酸烷基取代罗丹明螺硫酯和超干n,n-二甲基甲酰胺(dmf)的质量体积比为1.0:3.0-5.0(mg:ml)。本发明的有益效果:
本发明涉及的自闪烁罗丹明螺硫酯荧光染料具有合成原料低价、方法简单、易分离等优点。
本发明涉及的自闪烁罗丹明螺硫酯荧光染料在活细胞中具有良好的自闪烁性能,适用于活细胞随机光学重构显微成像。
本发明涉及的自闪烁罗丹明螺硫酯荧光染料通过蛋白标签技术连接到活细胞的目标区域并对其进行超分辨成像,其特异性好、定位准确,在hela细胞中的细胞核、线粒体和骨架tublin均可得到高分辨率的荧光成像图。
附图说明:
图1:为中间体1的氢核磁谱图。
图2:为中间体2的氢核磁谱图。
图3:为中间体3的氢核磁谱图。
图4:为中间体4的氢核磁谱图。
图5:为中间体5的氢核磁谱图。
图6:为中间体6的氢核磁谱图。
图7:为p1(10-5m)在不同溶剂中的紫外-可见吸收光谱。
图8:为p1(10-5m)在不同溶剂中的荧光发射光谱。
图9:为不同ph值条件下,p1(10-5m)水溶液的紫外-可见吸收光谱。
图10:为不同ph值条件下,p1(10-5m)水溶液的荧光发射光谱。
图11:为p1(10-5m)pkcycle的结果统计图,从图上可以看出在生理酸度范围内该自闪烁罗丹明螺硫酯荧光染料主要以闭环不发光的形式存在。
图12:为p1(10-5m)的hela细胞细胞核的storm重构图像。
图13:为p1(10-5m)的hela细胞线粒体的storm重构图像。
图14:为p1(10-5m)的hela细胞骨架tublin的storm重构图像。
具体实施方式
以下结合具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
实施例1
当r1=r2=ch2ch3,n=1时,一种自闪烁罗丹明螺硫酯荧光染料p1的合成路线如下:
其中,
中间体1的合成:
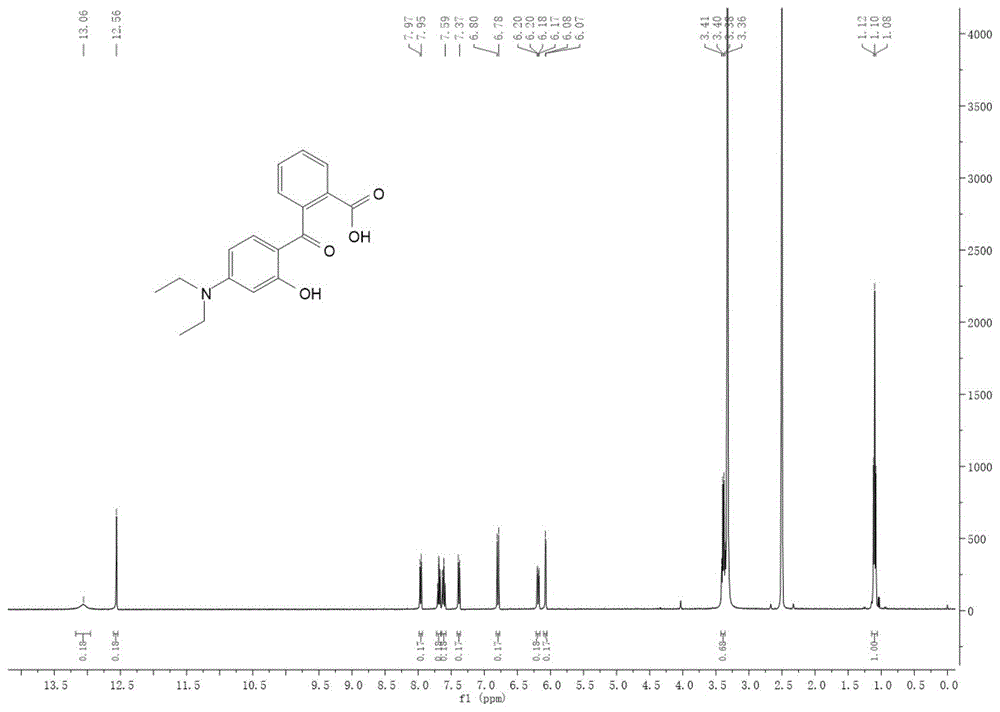
将3-羟基-n,n-二乙基苯胺(2.9037g,17.6mmol)和邻苯二甲酸酐(2.6319g,17.8mmol)置于100ml两口烧瓶中,抽真空并用氮气置换三次。在氮气保护下,用注射器向烧瓶中加入20ml甲苯,溶液呈黑色,加热回流16h,溶液由黑色变为紫红色。将反应液倒入100ml水中,抽滤,用少量甲醇洗涤滤饼,得到淡粉色固体。将得到的固体用异丙醇进行重结晶,重结晶析出的固体用少量甲醇洗涤,得到淡黄色固体(3.407g,产率61.8%)。
其高分辨质谱数据如下:
高分辨质谱c18h20no4+[m+h]+计算值:314.1392;实验值:314.1395。
其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ13.06(s,1h),12.56(s,1h),7.96(d,j=7.7hz,1h),7.69(td,j=7.5,1.2hz,1h),7.61(td,j=7.6,1.3hz,1h),7.38(d,j=7.5hz,1h),6.82–6.77(m,1h),6.19(dd,j=9.2,2.4hz,1h),6.07(d,j=2.4hz,1h),3.39(q,4h),1.10(t,j=7.0hz,6h)。
经鉴定,其产物结构为中间体1,如图1所示.
中间体2的合成:
将中间体1(3.1123g,9.94mmol)和间羟基苯基哌嗪(1.7840g,10.02mmol)置于100ml两口烧瓶中,抽真空并用氮气置换3次。在氮气保护下用注射器向烧瓶中注入甲磺酸(15ml,0.23mol)。在90℃下反应23h,溶液由黑色变为红黑色。
称取无水碳酸钠(13.6295g,0.13mmol)溶于约100ml水中,在冰水浴和搅拌条件下,缓慢将冷却后的反应液加入碳酸钠水溶液中,用ph试纸检测溶液呈弱碱性。用二氯甲烷对反应混合液进行萃取,上层为水相,下层为有机相,萃取过程中水相逐渐由红黑色变为淡粉色。合并后的有机相用无水硫酸钠进行干燥。用旋转蒸发仪除去溶剂后,剩余物用硅胶柱层析进行提纯(二氯甲烷/甲醇,10:1,v/v),得到红色固体(3.5763g,产率79.0%)。
其高分辨质谱数据如下:
高分辨质谱c28h30n3o3+[m]+计算值:456.2282;实验值:456.2279。
其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ7.98(d,j=7.6hz,1h),7.82–7.76(m,1h),7.71(td,j=7.5,0.7hz,1h),7.26(d,j=7.6hz,1h),6.76(d,j=2.3hz,1h),6.71(dd,j=8.9,2.5hz,1h),6.51(d,j=8.8hz,1h),6.44(q,j=8.6,1.8hz,3h),3.40–3.36(m,4h),3.20(t,5h),2.90(t,4h),1.10(t,j=7.0hz,6h)。
经鉴定,其产物结构为中间体2,如图2所示。
中间体3的合成:
将中间体2(3.4602g,7.59mmol)、芴甲氧羰酰琥珀酰亚胺(2.5571g,7.59mmol)和无水碳酸钠(1.064g,10.04mmol)置于100ml两口烧瓶中,抽真空并用氮气置换3次。在氮气保护下用注射器注入15ml乙腈,在室温下反应9.5h。用旋转蒸发仪除去溶剂后,用乙酸乙酯将粗产物溶解,抽滤,取滤液。用少量水洗涤滤液,有机相用无水硫酸钠进行干燥。除去溶剂后,剩余物用硅胶柱层析进行提纯(二氯甲烷/甲醇,20:1,v/v),得到粉色固体(4.3688g,产率85.0%)。
其高分辨质谱数据如下:
高分辨质谱c43h40n3o5[m+h]+计算值:678.2968;实验值:678.2971。
其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ7.99(d,j=7.6hz,1h),7.91(d,j=7.4hz,2h),7.79(t,j=7.5hz,1h),7.72(t,j=7.4hz,1h),7.65(d,j=7.4hz,2h),7.43(t,j=7.4hz,2h),7.35(t,j=7.4hz,2h),7.27(d,j=7.7hz,1h),6.78(s,1h),6.73(d,j=8.9hz,1h),6.54(d,j=8.8hz,1h),6.46(d,j=7.1hz,3h),4.43(d,j=6.4hz,2h),4.31(t,j=6.4hz,1h),3.45(s,4h),3.37(q,j=6.9hz,4h),3.17(s,4h),1.11(t,j=6.9hz,6h)。经鉴定,其产物结构为中间体3。
经鉴定,其产物结构为中间体3,如图3所示。
中间体4的合成:
将中间体3(4.3540g,6.43mmol)置于250ml圆底烧瓶中,加入30ml1,2-二氯乙烷使反应物完全溶解,溶液呈亮红色。在搅拌条件下向溶液中滴加三氯氧磷(1.5ml,16.125mmol),回流反应8h。用旋转蒸发仪除去反应混合物中的溶剂,剩余物用80ml四氢呋喃溶解,并将其缓慢滴加进含有硫脲(0.7748,10.19mmol)、10ml四氢呋喃、三乙胺(6ml,43.24mmol)和800μl水的混合溶液中,滴加过程中有白雾生成,且溶液变为暗紫色,烧瓶底部有白色沉淀。在室温下搅拌18.5h后除去溶剂,剩余物加入100ml水中,用乙酸乙酯对水相萃取,上层为有机相,下层为水相,有机相为淡红色。合并后的有机相用无水硫酸钠干燥。之后除去溶剂,粗产物用硅胶柱层析进行提纯(石油醚/乙酸乙酯,5:1,v/v),得到白色固体(2.4193g,产率54%)。其高分辨质谱数据如下:
高分辨质谱c43h40n3o5s[m+h]+计算值:694.2740;实验值:694.2886。其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ7.90(d,j=7.4hz,2h),7.83(d,j=7.7hz,1h),7.71(t,j=7.2hz,1h),7.62(dd,j=17.7,7.4hz,3h),7.42(t,j=7.3hz,2h),7.35(t,j=7.2hz,2h),7.18(d,j=7.6hz,1h),6.67(d,j=9.9hz,3h),6.62(d,j=9.0hz,1h),6.41(d,j=8.4hz,1h),6.32(s,1h),4.41(d,j=6.3hz,2h),4.30(t,j=6.2hz,1h),3.47–3.42(m,4h),3.12(s,4h),1.08(dd,j=11.3,6.8hz,6h)。
经鉴定,其产物结构为中间体4,如图4所示。
中间体5的合成:
将中间体4(2.3095g,3.3mmol)置于250ml圆底烧瓶中,向烧瓶中加入100ml15%的二乙胺乙腈溶液,在室温下搅拌5h。除去溶剂,剩余物用硅胶柱层析进行提纯(二氯甲烷/甲醇,20:1,v/v),得到紫色固体(0.8979g,产率56.5%)。
其高分辨质谱数据如下:
高分辨质谱c28h30n3o2s[m+h]+计算值:472.2059;实验值:472.2052。其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ7.82(d,j=7.5hz,1h),7.69(t,1h),7.60(t,j=7.1hz,1h),7.17(d,j=7.8hz,1h),6.65(s,2h),6.63–6.58(m,2h),6.40(dd,j=9.0,2.5hz,1h),6.31(d,j=2.5hz,1h),5.76(s,1h),3.30(d,j=6.9hz,4h),3.13–3.01(m,4h),2.85–2.77(m,4h),1.08(t,j=7.0hz,6h)。
经鉴定,其产物结构为中间体5,如图5所示。
中间体6的合成:
将中间体5(0.4382g,0.93mmol)、3-溴丙酸(0.1521g,0.99mmol)、无水碳酸钾(0.1417g,1.02mmol)和碘化钾(0.1759g,1.06mmol)置于100ml圆底烧瓶中,加入20ml乙腈使固体溶解,加热回流23h。反应后的溶液用旋转蒸发仪除去溶剂,剩余物用硅胶柱层析进行提纯(二氯甲烷/甲醇,10:1,v/v),得到紫色固体(0.2207g,产率44.1%)。
其高分辨质谱数据如下:
高分辨质谱c31h34n3o4s[m+h]+计算值:544.2270;实验值:544.2263。其核磁谱图氢谱数据如下:
1hnmr(400mhz,dmso)δ7.82(d,j=7.6hz,1h),7.70(t,j=7.0hz,1h),7.60(t,j=7.3hz,1h),7.17(d,j=7.7hz,1h),6.66(s,1h),6.64–6.58(m,2h),6.40(dd,j=8.9,2.3hz,1h),6.31(d,j=2.3hz,1h),3.33(s,4h),3.31(s,4h),3.16(s,4h),2.60(t,j=7.0hz,2h),2.41(t,j=7.0hz,2h),1.08(t,j=6.9hz,6h)。
经鉴定,其产物结构为中间体6,如图6所示。
p1的合成:
将中间体6(0.1822g,0.34mmol),1-羟基苯并三唑(hobt)(0.2255g,1.61mmol)和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc-hcl)(0.2859g,1.49mmol)置于50ml圆底烧瓶中,加入5ml超干n,n-二甲基甲酰胺,在搅拌条件下加入halo-nh2(0.1692g,0.76mmol)和0.34ml三乙胺,在室温下搅拌43h。除去反应液中的溶剂后,剩余物用硅胶柱层析进行提纯(二氯甲烷/甲醇,50:1,v/v),得到无色粘稠液体(0.03g,产率15%)。
其高分辨质谱数据如下:
高分辨质谱c41h54n4o5scl[m+h]+计算值:749.3503;实验值:749.3510。
经鉴定,其产物结构为化合物p1。
实施例2
将p1用dmso溶解,配制成浓度为1mm的测试母液。选取甲苯、四氢呋喃、1,4-二氧六环、乙酸乙酯、三氯甲烷、乙腈、n,n-二甲基甲酰胺、二甲基亚砜、甲醇和水作为测试用溶剂,稀释至终浓度为10-5m,分别测试其紫外-可见吸收光谱和荧光发射光谱,以考察其基态和激发态对溶剂环境的敏感性。如图7和8所示,实验结果表明,该分子基态和激发态均对溶剂环境不敏感,可尝试进一步应用到细胞内。
实施例3
将p1的1mm母液用盐酸和氢氧化钠的稀溶液分别配制成不同ph值且终浓度为10-5m的水溶液,然后分别测试其紫外-可见吸收光谱和荧光发射光谱,计算pkcycl值,以考察其是否可在生理酸度下保持大部分处于闭环不发光的状态。如图9、10和11所示,实验结果表明,该分子在ph7.4的生理条件下,其分子大部分处于闭环不发光的状态,是满足storm超分辨成像的必要条件之一。
实施例4
p1对活细胞不同细胞器染色后的荧光成像图。取p1的母液用1ml培养基稀释至终浓度为10-6m,然后将上述培养基加入到细胞培养皿中,37℃、5%co2下孵育10分钟后分别进行storm成像。
p1终浓度为10-6m的细胞培养液孵育转染了halo蛋白质粒到hela细胞细胞核的storm成像,如图12所示,hela细胞的细胞核清晰可见。
p1终浓度为10-6m的细胞培养液孵育转染了halo蛋白质粒到hela细胞线粒体的storm成像,如图13所示,hela细胞的线粒体清晰可见。
p1终浓度为10-6m的细胞培养液孵育转染了halo蛋白质粒到hela细胞骨架tublin的storm成像,如图14所示,hela细胞的骨架清晰可见。
- 还没有人留言评论。精彩留言会获得点赞!