基于免标记的荧光探针构建的FEN1酶活性检测方法及其应用与流程
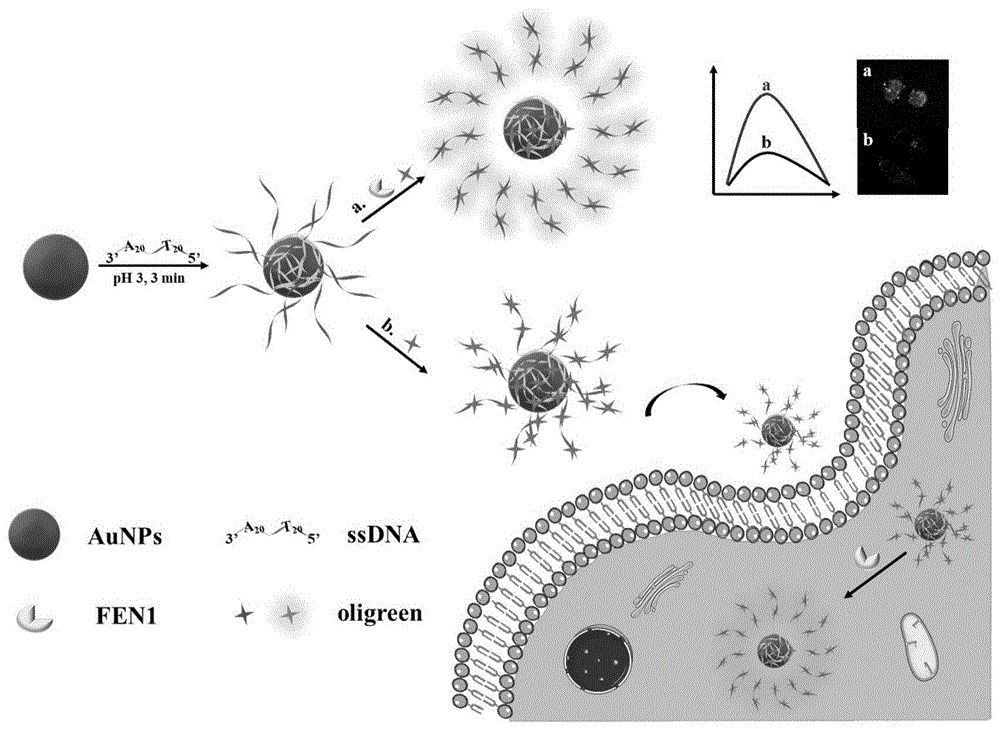
本发明属于生物技术领域,具体涉及基于免标记的荧光探针构建的fen1酶活性的检测方法及其应用。
背景技术:
瓣状核酸内切酶(fen1)是一种金属依赖性核酸酶,它只识别特定的结构而不依赖于序列,主要切除双链dna中悬挂的5’-端序列。在生物体内参与dna复制和损伤修复过程,主要涉及冈崎片段成熟和长片段碱基切除修复。然而,过表达的fen1酶会促进肿瘤细胞的增值、分化。研究表明,相比于正常组织,肿瘤组织内的fen1酶含量更高,而且,过表达的fen1酶与肿瘤的大小、分化程度以及tnm分期有关。因此,fen1酶可以作为一种新型的肿瘤标志物,通过检测生物体内fen1酶的含量来预判肿瘤的发生、发展。
在过去的几十年里,fen1酶曾作为辅助酶广泛应用于生物传感器的构建,主要起到信号循环放大的作用。但是,把fen1酶作为检测对象的研究却不是很多,尤其是定量分析检测。尽管困难重重,我们还是构建出一种免标记的荧光探针对fen1酶进行定量检测,而且进行了细胞成像。
与传统的酶联免疫吸附测定(elisa)方法相比,荧光方法操作更为简单,检测灵敏度更高,而且免标记省去了许多繁琐的标记步骤。因此,免标记的荧光探针的构建更利于fen1酶活性的定量检测。
技术实现要素:
发明目的:本发明所要解决的技术问题是提供了一种基于免标记的荧光探针及其制备方法和应用。
本发明还要解决的技术问题是还提供了基于免标记的荧光探针构建的fen1酶活性的检测方法及在细胞成像中的应用。本发明不需要复杂的标记过程,简化了检测方法,极大地降低了fen1酶检测成本,具有运行成本低、检测快速、简便、选择性好等优点。此外,所制备的免标记的荧光探针具有较好的生物相容性和低毒性特点,可以进入细胞,进行体内检测。
技术方案:为了解决上述技术问题,本发明提供了免标记的荧光探针的制备方法,所述免标记的荧光探针的制备方法如下:将金纳米粒子与ssdna溶液混合后涡旋5~10秒得到混合溶液,在混合溶液加入柠檬酸缓冲溶液,3~5分钟后加入hepes缓冲溶液,离心洗去多余ssdna即得。
其中,所述金纳米粒子浓度为10~12nm,ssdna浓度为100nm。
其中,所述柠檬酸缓冲溶液为500mm柠檬酸,ph2.5~3.5;hepes缓冲溶液为500mmhepes,ph6.5~7.5。
本发明内容还包括所述的制备方法制备得到的免标记的荧光探针。
本发明内容还包括所述的基于免标记的荧光探针构建的fen1酶活性检测方法,所述检测方法包括以下步骤:将所述的免标记的荧光探针与酶反应缓冲溶液混合,然后加入fen1酶,最后再加入荧光信号分子,保持黑暗环境,在室温下反应一段时间后得到溶液,记录溶液的荧光光谱,通过荧光强度得出fen1酶的浓度。
其中,所述荧光探针与酶反应缓冲溶液的体积比0.1~1。
其中,所述荧光信号分子为oligreen溶液,所述oligreen溶液稀释倍数为0.5~1x104倍。
其中,所述酶反应缓冲溶液为50mmtris-hcl、10mmmgcl2,ph6~8。
本发明内容还包括所述的免标记的荧光探针、所述的fen1酶活性检测方法在细胞成像中的应用。
其中,所述细胞含有fen1酶的任何细胞。
具体的,所述的细胞可以但不仅限于正常肝脏细胞(lo2),肝脏癌细胞(a549和hepg2)和乳腺癌细胞(mcf-7)等。
具体地,本发明所述荧光探针的合成:100μl的13nm的金纳米粒子与2μl100nm的ssdna混合均匀后静置3分钟;然后加入2μlph3.0的柠檬酸缓冲溶液,混合均匀后静置3分钟,最后加入6μlhepes缓冲溶液,使混合溶液恢复中性环境。最后离心去除多余的ssdna(12000rpm,10分钟)。保留沉淀,复溶后进行后续实验。
具体地,不同浓度fen1酶检测步骤及标准曲线获得:将含有5μl制备的免标记的荧光探针、50μl酶反应缓冲溶液和不同浓度的fen1酶混合均匀后在37℃温育2小时。接下来,将5μloligreen溶液(0.005x)和tris-hcl缓冲溶液在黑暗环境下室温反应15分钟,最终溶液体积为200μl。在室温下记录荧光光谱,得出荧光强度与不同浓度fen1酶的校准曲线。
本发明通过不同浓度fen1酶的校准曲线,采用同样的检测步骤获得待测的fen1酶浓度。
其中,所述酶缓冲溶液为50mmtris-hcl、10mmmgcl2,ph6~8。
有益效果:与现有技术相比,本发明具有如下的特色及优点:本发明原理简单、实验周期短、所用原料成本较低,无需任何大型仪器,在相同条件下可以检测出更低含量的待测物。oligreen与ssdna结合后会产生荧光,但是由于距金纳米粒子表面较近,在没有fen1酶存在的条件下,产生的荧光会被金纳米粒子猝灭。在有fen1酶存在的条件下,fen1酶会将悬挂的5’-端dna序列切除,使其远离金纳米粒子表面,得到较强的荧光信号。本发明不需要借助昂贵精密仪器检测,简化了检测方法,极大地降低了fen1酶检测成本,本发明具有运行成本低、检测快速简便、选择性好、灵敏度高等优点。此外,所制备的荧光探针具有较好的生物相容性和低毒性特点,可以进入细胞,进行体内检测。
附图说明
图1显示了基于免标记的荧光探针构建的fen1酶活性的检测及细胞成像分析方法的流程图;
图2显示了基于免标记的荧光探针构建的fen1酶活性的检测及细胞成像分析方法的原理图;(a)aunps-ssdna,(b)oligreen,(c)oligreen/aunps,(d)oligreen/aunps-ssdna,(e)oligreen/fen1/aunps-ssdna
图3a显示了金纳米粒子的透射电子显微镜(tem)图像;图3b显示了金纳米粒子的粒径分布图像;图3c显示了金纳米粒子和荧光探针的紫外吸收光谱图;图3d显示了金纳米粒子和荧光探针的zeta电位图;
图4a显示了在不同浓度fen1酶浓度(a-j分别对应浓度为0u、0.05u、0.1u、0.25u、0.5u、0.75u、1.0u、1.5u、2u、4u)作用下得到的荧光光谱图;图4b显示了荧光强度与不同浓度fen1酶的校准曲线。(插图:荧光强度与fen1酶对数浓度之间的线性关系,范围为0.05u~2u);
图5显示了免标记的荧光探针和oligreen溶液对不同细胞的毒性;横坐标柱状图从左至右依次为lo2细胞,mcf-7细胞,hepg2细胞和a549细胞。
图6显示了不同细胞内检测fen1酶的共聚焦图像。
具体实施方式
下面通过具体的实施例和附图对本发明进一步说明,应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干变型和改进,这些也应视为属于本发明的保护范围。
本实验中用到的试剂和仪器:
四水合氯金酸(haucl4·4h2o)、二水合柠檬酸三钠(c6h5na3o7·2h2o)、柠檬酸、盐酸、三(羟甲基)氨基甲烷、六水合氯化镁以及盐酸购自国药化学试剂公司(中国上海)。2-[4-(2-羟乙基)哌嗪-1-基]乙磺酸(hepes)购自aladdinindustrialcorporation(中国上海)。瓣状核酸内切酶(fen1)购自newenglandbiolabs。胎牛血清,dmem培养基,1640培养基以及3-(4,5-二甲基噻唑-2-基)-2-二苯基溴化四唑(mtt)购自凯基生物(江苏南京)。oligreen溶液购自thermofisherscientific。dna链由生工生物(中国上海)提供。
ssdna:5’-ttttttttttttttttttttaaaaaaaaaaaaaaaaaaaa-3’
本发明实施例中的酶反应缓冲溶液为:50mmtris-hcl、10mmmgcl2,ph6~8。
本发明实施例中的tris-hcl缓冲溶液为5~10mmtris-hcl,ph6.5~8.5。
本发明实施例中的pbs缓冲溶液为:137mmnacl、10mmnah2po4、2.7mmkcl、2mmkh2po4,ph7.4。
实施例1金纳米粒子aunps的合成
首先,于洗净的双口圆底烧瓶内称取50g蒸馏水,加入1030μl2%的haucl4·2h2o,回流加热并搅拌(132℃,20000rpm);然后,称取0.057g柠檬酸三钠溶于5ml蒸馏水,所得溶液迅速加入到上述沸腾的氯金酸溶液中,继续加热15分钟后,停止加热,继续搅拌至溶液冷却到室温。所用圆底烧瓶,冷凝管在使用前需依次泡碱缸12h,王水(v盐酸∶v硝酸=3∶1)24h,最后用蒸馏水洗净得到金纳米粒子aunps备用。
实施例2荧光探针aunps-ssdna的合成
100μl的实施例1制备的13nm的金纳米粒子aunps与2μl100nm的ssdna混合均匀后静置3分钟;然后加入2μlph3.0的柠檬酸缓冲溶液,混合均匀后静置3分钟,最后加入6μlhepes缓冲溶液(500mm,ph7.0),使混合溶液恢复中性环境。最后离心去除多余的ssdna(12000rpm、10分钟)。保留沉淀,复溶后得到荧光探针aunps-ssdna进行后续实验。
图3a显示了合成的金纳米粒子aunps的tem图像,从图3a中可以看出,金纳米aunps粒子大小均一,分布均匀;图3b显示了金纳米粒子aunps的粒径分布图,表明金纳米粒子aunps的粒径主要分布在13nm;图3c是金纳米粒子aunps和荧光探针aunps-ssdna的紫外吸收谱图,可以看到金纳米粒子aunps在519nm处的吸收峰和dna链在260nm处的吸收峰;图3c是金纳米粒子aunps和荧光探针aunps-ssdna的zeta电位图,可以看出带负电的金纳米粒子在修饰上dna链后,电位变得更负。以上结果表明成功制备荧光探针。
实施例3荧光探针aunps-ssdna的合成
100μl的实施例1制备的13nm的金纳米粒子与2μl100nm的ssdna混合均匀后静置3分钟;然后加入2μlph3.0的柠檬酸缓冲溶液,混合均匀后静置5分钟,最后加入6μlhepes缓冲溶液,使混合溶液恢复中性环境。最后离心去除多余的ssdna(~12000rpm,10分钟)。保留沉淀,复溶后得到荧光探针aunps-ssdna进行后续实验。实施例4荧光探针aunps-ssdna的合成
100μl的实施例1制备的13nm的金纳米粒子与4μl100nm的ssdna混合均匀后静置3分钟;然后加入2μlph3.0的柠檬酸缓冲溶液,混合均匀后静置3分钟,最后加入6μlhepes缓冲溶液,使混合溶液恢复中性环境。最后离心去除多余的ssdna(~12000rpm,10分钟)。保留沉淀,复溶后得到荧光探针aunps-ssdna进行后续实验。实施例5不同浓度的fen1酶的检测
将含有5μl实施例2制备的荧光探针aunps-ssdna、50μl酶反应缓冲溶液和不同浓度(0u、0.05u、0.1u、0.25u、0.5u、0.75u、1.0u、1.5u、2u、4u)的fen1酶混合均匀后在37℃温育2小时。接下来,将5μloligreen溶液(0.005x)和tris-hcl缓冲溶液在黑暗环境下室温反应15分钟,最终溶液体积为200μl。实验结果图4a显示了定量检测fen1酶的荧光强的变化图;图4b显示了荧光强度与不同浓度fen1酶的校准曲线。(插图:荧光强度与fen1酶对数浓度之间的线性关系,范围为0.05u~2u,检测限为0.007u)。
实施例6荧光探针和oligreen溶液对不同细胞的毒性检测
分别将适宜密度(104个/孔)的细胞(lo2细胞,mcf-7细胞,hepg2细胞和a549细胞)接种在96孔板内,加入200μl培养基(lo2细胞和mcf-7细胞为1640培养基,hepg2细胞和a549细胞为dmen培养基),然后向孔板内加入实施例2制备的荧光探针aunps-ssdna和oligreen溶液(0.005x)各5μl,分别在37℃培养箱中孵育2小时,6小时,18小时,24小时,32小时和48小时;孵育结束后,吸出培养基,加入200μlmtt溶液(0.5mg/ml,由pbs溶液稀释),继续孵育4小时;最后吸出mtt溶液,加入150μldmso溶液,震荡5分钟后用酶标仪记录吸光度。如图5所示,在与荧光探针孵育48小时后,基本所有细胞都能保持80%以上的存活率,由此可以证明荧光探针和oligreen溶液的低毒性。
实施例7不同细胞内检测fen1酶的共聚焦图像
分别将适宜密度(105个)的细胞(lo2细胞,mcf-7细胞,hepg2细胞和a549细胞)接种在共聚焦小皿内,过夜,使其贴壁培养(lo2细胞和mcf-7细胞为1640培养基,hepg2细胞和a549细胞为dmen培养基);然后将实施例2制备的荧光探针aunps-ssdna和oligreen溶液(0.005x)各20μl,加入460μl培养基稀释后加入共聚焦小皿中,在培养箱中37℃继续孵育4小时;最后用pbs缓冲液洗去未被胞吞的荧光探针aunps-ssdna和oligreen溶液,进行共聚焦成像拍摄。
如图6所示,在正常肝脏细胞(lo2)内fen1酶含量低,所以拍摄到的荧光强度相对较弱,在肝脏癌细胞(a549和hepg2)和乳腺癌细胞(mcf-7)内fen1酶含量高,对应的荧光强度也较强。
实施例8fen1酶在不同细胞内的实际样品检测及加标回收
为例进一步研究此方法在实际样品中的应用,分别将正常细胞(lo2)和癌细胞(a549,hepg2,mcf-7)的细胞核和细胞质分别进行了提取,参照实施例5的所建立的检测方法对实际样品中的fen1酶含量进行检测。此外,我们还在实际样品中进行了加标回收实验,得到较好的回收率,验证了该实验的准确性。结果如表1所示,这里是每种细胞取500个进行了对比。
表1
这些结果表明该发明所提出来的方法具有良好的性能,可以用于实际样品的检测。
序列表
<110>东南大学
<120>基于免标记的荧光探针构建的fen1酶活性检测方法及其应用
<160>1
<170>siposequencelisting1.0
<210>1
<211>40
<212>dna
<213>ssdna(artificialsequence)
<400>1
ttttttttttttttttttttaaaaaaaaaaaaaaaaaaaa40
- 还没有人留言评论。精彩留言会获得点赞!