一种层析介质及其制备方法和应用与流程
本发明涉及一种层析介质及其制备方法,属于生物化工领域中的蛋白质层析分离技术领域。
背景技术:
抗体类药物是目前生物技术类药物中最重要的一类,因其专一性强、亲和力高,在医药和研究领域具有广泛的应用前景。如在自身免疫疾病、体外诊断和检测等方面的应用。这类药物在生物医药市场的规模已经超过20%,且逐年增加。随着市场需求和生产规模的不断扩大,如何从复杂的料液中快速分离纯化抗体成为不可回避的问题,这也对抗体分离技术提出了越来越高的要求。
通常情况下对抗体产品的纯度有极高的要求,且需要在其保持活性的情况下才能发挥药物特性,然而传统的分离方法很难满足这些要求。如传统的疏水作用色谱需要在盐浓度相对较高的情况下分离抗体,然而在高盐浓度下很容易引起抗体活性的下降甚至失去活性,且特异性和选择性较差,分离步骤多、纯化效果有限。目前工业上应用较多的是以蛋白a或蛋白g为配基的亲和层析技术,蛋白a或蛋白g能够与抗体的fc段特异性的结合,因而具有很高的选择性;但这类配基不仅价格昂贵,操作成本高,且易被蛋白酶降解脱落,重复使用次数低,不易灭菌处理,从而限制其大规模的应用。因此,开发具有抗体选择性、稳定性好、且成本较低的拟蛋白a类配基具有很高的应用价值。
目前已报道的针对抗体分离的非蛋白类配基很多,有仿生亲和配基cibraconbluefg-3(j.chromatogr.a,1979,165:301)、基于dna寡核苷酸的适配子,如6h5和6h5适配子(biotechnol.bioeng,2011,108(10):2371)和denano(nucleic.acids.res,2016,44(10):e96)等。这类配基主要强调对蛋白的特异性及亲和力,但不可避免的造成抗体洗脱条件苛刻或者回收率低。其次,有些仿生配基在洗脱过程中易脱落,而适配子的富集过程复杂、成本较高。
burton和harding等人在1998年首次提出了基于混合模式色谱的疏水电荷诱导层析技术(hydrophobicchargeinductionchromatography,hcic),通过调节缓冲液的ph,利用抗体与配基之间的静电排斥作用实现高效洗脱(j.chromatogr.a,1998,814:71)。yanjun等人提出了一种基于5-氨基苯并咪唑的疏水电荷诱导配基,用于从细胞培养上清中分离免疫球蛋白g(j.sep.sci.2015,38:2387)。中国发明专利申请公开说明书cn101279243和cn101278244也报道了在hcic介质的间隔臂上引入含硫基团来提高介质对抗体的选择性以及相关制备方法。这类介质能够在一定程度上满足抗体分离要求,且有些商品化的层析介质已具有一定规模的工业应用。但总体来看,现有的混合模式层析介质主要用于抗体聚集体的分离,还未能替代蛋白a用于抗体的精分离步骤,主要原因在于处理能力有限,吸附容量并没有得到有效的提高,难以在工业规模的抗体分离纯化过程中体现出优势。因此,通过优化和设计配基的结构,在满足抗体吸附选择性的基础上有效提高吸附介质的抗体载量,对于工业规模抗体的高效分离具有重要的意义。
技术实现要素:
为了解决现有层析介质的局限性,本发明将5位取代的苯并三唑作为功能配基,通过间隔臂固载在基质材料表面,可以用于疏水电荷诱导层析分离,具有非盐依赖吸附的特性。
本发明的技术方案为,一种层析介质,包括由间隔臂偶联的基质和功能配基,所述功能配基为5位取代的苯并三唑,结构为:
其中,r为羧基、氨基、巯基或羟基。
作为优选地,所述的基质为带有羟基的亲水性多孔微球。
作为优选地,所述基质的材料为交联琼脂糖凝胶、纤维素、硅胶或经亲水性改造的聚苯乙烯,也可以为领域内技术人员常用的其他形式的亲水性微球载体材料。
本发明所述的交联琼脂糖凝胶,优选琼脂糖凝胶与2,3-二溴丙醇反应而成的交联琼脂糖凝胶,以增强琼脂糖凝胶的物理化学稳定性。
所述的层析介质的配基密度为27-164μmol/ml干胶。优选配基密度大于100μmol/ml干胶,该密度范围内的凝胶介质的抗体动态结合容量更高。
本发明所述的干胶是指,湿胶状态的基质经砂芯漏斗抽滤后无水析出即为干胶。
作为优选地,所述的功能配基为5-羧基苯并三唑。
当功能配基为5-羧基苯并三唑时,优选地,所述的间隔臂为双氨基试剂。
本发明所述的双氨基试剂包括但不限于1,2-乙二胺、1,6-己二胺和3,3'-二氨基二丙基胺。
进一步优选地,当双氨基试剂为1,2-乙二胺时,所述的层析介质的结构为,
当双氨基试剂为1,6-己二胺时,所述的层析介质的结构为,
当双氨基试剂为3,3'-二氨基二丙基胺时,所述的层析介质的结构为,
作为优选地,所述的功能配基为5-氨基苯并三唑。
当功能配基为5-氨基苯并三唑时,优选地,所述的间隔臂为6-氨基己酸,所述的层析介质的结构为,
本发明还提供上述层析介质作为疏水电荷诱导层析模式分离和纯化抗体中的应用。
本发明还提供上述层析介质作为抗体选择性吸附剂在自身免疫性疾病的血液净化治疗中的应用。
本发明还提供上述以5-羧基苯并三唑为功能配基的层析介质的制备方法,包括如下步骤:
⑴活化
基质经去离子水、丙酮洗涤后,以丙酮为溶剂,以n,n'-羰基二咪唑为活化剂,25~37℃下震荡反应2~5小时;反应物的加入量,以体积份计,基质:丙酮=1:2~4;以质量份计,基质:n,n'-羰基二咪唑=1:0.01~0.1;活化后使用丙酮洗涤,得活化的基质;
⑵接枝间隔臂
将步骤⑴所得活化的基质与丙酮、双氨基试剂混合,在30-37℃下震荡反应5-8小时;反应物的加入量,以体积份计,丙酮为步骤⑴基质的2~4倍;以质量份计,双氨基试剂为步骤⑴基质的0.1~0.3倍;反应后用丙酮洗涤,得接枝间隔臂的基质;
⑶偶联功能配基
以n,n'-二甲基甲酰胺为溶剂,将步骤⑵所得的接枝间隔臂的基质与n,n'-二异丙基碳二酰亚胺、所述配基混合,30-37℃震荡反应12~24小时;反应物的加入量,以体积份计,n,n'-二甲基甲酰胺为步骤⑴基质的2~4倍;以质量份计,n,n'-二异丙基碳二酰亚胺为步骤⑴基质的0.1~0.3倍,以摩尔比计,n,n'-二异丙基碳二酰亚胺:配基=1:0.3;反应后依次用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤。
本发明还提供上述5-氨基苯并三唑为功能配基的层析介质的制备方法,包括如下步骤:
⑴活化
基质经去离子水、丙酮洗涤后,以丙酮为溶剂,以n,n'-羰基二咪唑为活化剂,25~37℃下震荡反应2~5小时;反应物的加入量,以体积份计,基质:丙酮=1:2~4;以质量份计,基质:n,n'-羰基二咪唑=1:0.01~0.1;活化后使用丙酮洗涤,得活化的基质;
⑵接枝间隔臂
将步骤⑴所得活化的基质与丙酮、6-氨基己酸混合,在30-37℃下震荡反应5-8小时;反应物的加入量:以体积份计,丙酮的为步骤⑴基质的2~4倍;以质量份计,6-氨基己酸为步骤⑴基质的0.1~0.3倍;反应后用丙酮洗涤,得接枝6-氨基己酸的基质;
⑶偶联功能配基
以n,n'-二甲基甲酰胺为溶剂,将步骤⑵所得的接枝6-氨基己酸的基质与n,n'-二异丙基碳二酰亚胺、所述配基混合,30-37℃震荡反应12~24小时;反应物的加入量:以体积份计,n,n'-二甲基甲酰胺为步骤⑴基质的2~4倍;以质量份计,n,n'-二异丙基碳二酰亚胺为步骤⑴基质的0.1~0.3倍;以摩尔比计,n,n'-二异丙基碳二酰亚胺:配基=1:0.3,反应后依次用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤。
本发明研制的以5位取代的苯并三唑为功能配基的层析介质,制备过程简单、成本低,可以用于疏水电荷诱导层析分离和纯化抗体,也可作为抗体选择性吸附剂用于自身免疫性疾病的血液净化治疗,具有以下优点:
(1)抗体吸附容量大。该配基以苯并三唑为结构主体,包含芳香环和五元杂环,疏水程度适当,对蛋白具有显著的吸附能力,结合速率快;相对于其他类型抗体吸附介质具有高的动态结合容量;
(2)吸附过程几乎不受盐浓度的影响,因此无需对料液进行预处理,可直接捕获目标抗体,减少分离过程中的操作步骤;
(3)洗脱方便,苯并三唑配基的酸解离常数(pka)较高,容易质子化,只需要将洗脱液ph调节到弱酸性,即可实现高效洗脱,避免了过酸、过碱及高盐等对抗体的活性造成损失;
(4)层析介质稳定性高,苯并三唑功能化的层析介质在强碱、强酸中浸泡,及经过高温灭菌后,依旧保持很好的稳定性;
(5)介质非特异性吸附小,通过调节上样及洗脱缓冲液的ph,可有效地减少层析介质的非特异性吸,提高了对抗体的吸附选择性。
附图说明
本发明附图6幅,
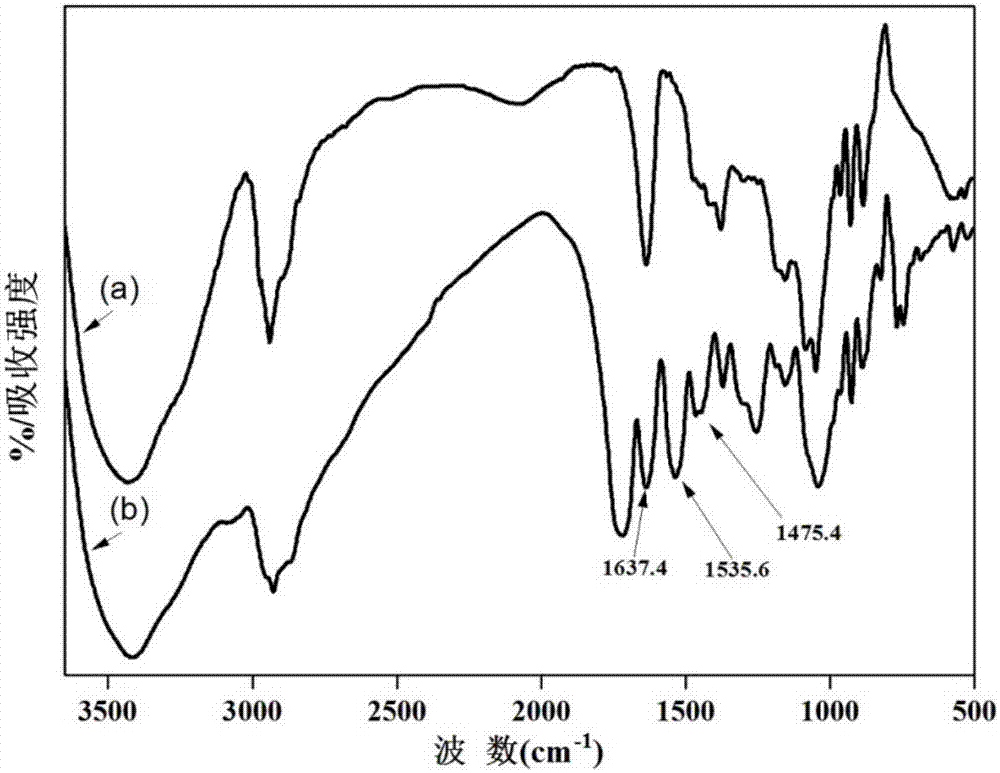
图1为5-羧基苯并三唑功能化交联琼脂糖凝胶红外光谱图,图1中,a.交联琼脂糖凝胶;b.偶联5-羧基苯并三唑的交联琼脂糖凝胶;
图2为5-氨基苯并三唑功能化交联琼脂糖凝胶红外光谱图,图2中,a.交联琼脂糖凝胶;b.偶联5-氨基苯并三唑的交联琼脂糖凝胶;
图3不同溶液ph下的抗体静态平衡吸附容量图;
图4不同ph洗脱下蛋白成分电泳图,图4中,1:marker;2:人血浆;3:流穿;4:ph4.0洗脱;5:ph3.0洗脱;
图5氯化钠浓度对抗体动态结合容量的影响;
图6苛性条件处理对功能化交联琼脂糖凝胶吸附抗体的影响。
具体实施方式
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
实施例1利用3,3'-二氨基二丙基胺作为间隔臂偶联5-羧基苯并三唑
⑴活化
取交联琼脂糖凝胶(gehealthcare,sepharosetmcl-6b)10g,用去离子水多次润洗,再使用适量丙酮洗涤3~5次,抽干后加入到含有0.1~1.0g的n,n'-羰基二咪唑的20~40ml丙酮溶液中,在摇床中25~37℃、150~200rpm活化2~5小时,活化后使用丙酮洗涤,抽滤后,得活化的交联琼脂糖凝胶;
⑵接枝间隔臂
将活化的交联琼脂糖凝胶和1~3g的3,3'-二氨基二丙基胺加入到20~40ml丙酮溶液中,在摇床中30~37℃、150~200rpm反应5~8小时,反应后使用丙酮洗涤,除去未反应的3,3'-二氨基二丙基胺,抽干,得末端接枝3,3'-二氨基二丙基胺作为间隔臂的交联琼脂糖凝胶;
⑶偶联功能配基
将末端接枝有3,3'-二氨基二丙基胺作为间隔臂的交联琼脂糖凝胶和1~3gn,n'-二异丙基碳二酰亚胺,0.388~1.163g5-羧基苯并三唑的加入到20~40ml的n,n'-二甲基甲酰胺中,在摇床中30~37℃、150~200rpm反应12-24小时,反应后依次使用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤,除去未反应的5-羧基苯并三唑,得到以3,3'-二氨基二丙基胺为间隔臂的5-羧基苯并三唑功能化交联琼脂糖凝胶,配基密度为27-164μmol/ml干胶。附图1显示了偶联5-羧基苯并三唑的交联琼脂糖凝胶的红外光谱图,证明5-羧基苯并三唑作为配基已成功偶联到交联琼脂糖凝胶表面。
实施例2利用1,6-己二胺作为间隔臂偶联配基
⑴活化
取交联琼脂糖凝胶(gehealthcare,sepharosetmcl-6b)10g,用去离子水多次润洗以除去20%乙醇,再使用适量丙酮洗涤3~5次,抽干后加入到含有0.1~1.0g的n,n'-羰基二咪唑的20~40ml丙酮溶液中,在摇床中25~37℃、150~200rpm活化2~5小时,活化后使用丙酮洗涤,除去未反应的n,n'-羰基二咪唑,抽滤后,得活化的交联琼脂糖凝胶;
⑵接枝间隔臂
将活化的交联琼脂糖凝胶和1~3g的1,6-己二胺加入到20~40ml丙酮的溶液中,在摇床中30~37℃、150~200rpm反应5~8小时,反应后使用丙酮洗涤,除去未反应的1,6-己二胺,抽干,得末端接枝1,6-己二胺作为间隔臂的的交联琼脂糖凝胶;
⑶偶联功能配基
将末端接枝有1,6-己二胺作为间隔臂的交联琼脂糖凝胶、1~3gn,n'-二异丙基碳二酰亚胺和0.388~1.163g的5-羧基苯并三唑加入到20~40ml的n,n'-二甲基甲酰胺中,,30~37℃下150~200rpm摇床中反应12~24小时,反应后依次使用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤,除去未反应的5-羧基苯并三唑,得到以1,6-己二胺为间隔臂的5-羧基苯并三唑功能化交联琼脂糖凝胶,配基密度为31~157μmol/ml干胶。
实施例3利用1,2-乙二胺作为间隔臂偶联配基
⑴活化
取交联琼脂糖凝胶(gehealthcare,sepharosetmcl-6b)10g,用去离子水多次润洗除去20%乙醇,再使用适量丙酮洗涤3~5次,抽干后加入到含有0.1~1.0g的n,n'-羰基二咪唑的20~40ml丙酮溶液中,在摇床中25-37℃、150~200rpm活化2~5小时,活化后使用丙酮洗涤,除去未反应的n,n'-羰基二咪唑,抽滤后,得活化的交联琼脂糖凝胶;
⑵接枝间隔臂
将活化的交联琼脂糖凝胶和1~3g的1,2-乙二胺加入到20~40ml丙酮溶液中,30~37℃下150~200rpm摇床中反应5~8小时,反应后使用丙酮洗涤,除去未反应的1,2-乙二胺,抽干,得末端接枝有1,2-乙二胺作为间隔臂的交联琼脂糖凝胶;
⑶偶联功能配基
将末端接枝有1,2-乙二胺作为间隔臂的交联琼脂糖凝胶、1~3g的n,n'-二异丙基碳二酰亚胺和0.388~1.163g的5-羧基苯并三唑加入到20~40mln,n'-二甲基甲酰胺中,在摇床中30~37℃、150~200rpm反应12~24小时,反应后依次使用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤,除去未反应的5-羧基苯并三唑,得到以1,2-乙二胺为间隔臂的5-羧基苯并三唑功能化交联琼脂糖凝胶,配基密度为28~161μmol/ml干胶。
实施例4利用6-氨基己酸作为间隔臂偶联5-氨基苯并三唑
⑴活化
取交联琼脂糖凝胶(gehealthcare,sepharosetmcl-6b)10g,用去离子水多次润洗以除去20%乙醇,再使用适量丙酮洗涤3~5次,抽干后加入到含有0.1~1.0g的n,n'-羰基二咪唑的20~40ml丙酮溶液中,在摇床中25~37℃、150~200rpm活化2~5小时,活化后使用丙酮洗涤,除去未反应的n,n'-羰基二咪唑,抽滤后,得活化的交联琼脂糖凝胶;
⑵接枝间隔臂
将活化的交联琼脂糖凝胶和1~3g的6-氨基己酸加入到20~40ml丙酮溶液中,在摇床中30~37℃、150~200rpm反应5~8小时,反应后使用丙酮洗涤,除去未反应的6-氨基己酸,抽干,得末端接枝6-氨基己酸作为间隔臂的的交联琼脂糖凝胶;
⑶偶联5-氨基苯并三唑
将末端接枝有6-氨基己酸作为间隔臂的交联琼脂糖凝胶、1~3g的n,n'-二异丙基碳二酰亚胺和0.319~0.957g的5-氨基苯并三唑加入到20~40mln,n'-二甲基甲酰胺中,,30~37℃下150~200rpm摇床中反应12~24小时,反应后依次使用n,n'-二甲基甲酰胺、丙酮和碱性溶液(0.05mol/l氢氧化钠溶液)洗涤,除去未反应的5-氨基苯并三唑,得到以6-氨基己酸为间隔臂的5-氨基苯并三唑功能化交联琼脂糖凝胶,配基密度为28~162μmol/ml干胶。附图2显示了偶联5-氨基苯并三唑的交联琼脂糖凝胶的红外光谱图,证明5-氨基苯并三唑作为配基已成功偶联到交联琼脂糖凝胶表面。附图2中对应基团吸收峰位置与附图1中出峰位置略有差异,原因是由于苯环上偶联的羰基变成氨基导致特征吸收峰偏移。
实施例5利用6-氨基己酸为间隔臂偶联甲酸
⑴活化
取交联琼脂糖凝胶(gehealthcare,sepharosetmcl-6b)10g,用去离子水多次润洗以除去20%乙醇,再使用适量丙酮洗涤3~5次,抽干后加入到含有0.1~1.0g的n,n'-羰基二咪唑的20~40ml丙酮溶液中,在摇床中25~37℃、150~200rpm活化2~5小时,活化后使用丙酮洗涤,除去未反应的n,n'-羰基二咪唑,抽滤后,得活化的交联琼脂糖凝胶;
⑵接枝间隔臂
将活化的交联琼脂糖凝胶和1~3g的6-氨基己酸加入到20~40ml丙酮溶液中,在摇床中30~37℃、150~200rpm反应5~8小时,反应后使用丙酮洗涤,除去未反应的6-氨基己酸,抽干,得末端接枝6-氨基己酸作为间隔臂的的交联琼脂糖凝胶;
⑶偶联功能配基
将末端接枝有6-氨基己酸作为间隔臂的交联琼脂糖凝胶、1~3g的n,n'-二异丙基碳二酰亚胺和0.109~0.328g的甲酸,加入到20~40mln,n'-二甲基甲酰胺中,30~37℃下150~200rpm摇床中反应24~36小时,直至经茚三铜显色反应检测无初级胺剩余。反应后依次使用n,n'-二甲基甲酰胺、丙酮和去离子水洗涤,除去未反应的甲酸,得到以6-氨基己酸为间隔臂的甲酸封闭的交联琼脂糖凝胶。该功能化的交联琼脂糖凝胶用来考查当苯并三唑配基被一个氢原子替换后,在不同密度下对抗体的动态吸附行为。
实施例6静态饱和吸附容量的测定
采用实施例1中配基密度为164μmol/ml的交联琼脂糖凝胶7份,每份质量为0.08g,加入到5ml离心管中,并分别加入浓度为0.4、0.8、1.2、2.0、5.0、10.0、20.0的igg溶液2.0ml,封口后置于摇床中,28℃、130rpm吸附2h。吸附结束后,取上述混合液的上清液,用紫外分光光度计测定280nm下吸光值,按照1.0mg/mligg在280nm下的吸光值近似等于1.4换算,测定上清液中igg浓度,并按照质量守恒计算单位体积功能化交联琼脂糖凝胶对igg的结合量(交联琼脂糖凝胶的质量体积比为0.8,即0.8g交联琼脂糖凝胶体积约1.0ml),上述实验每组重复3次,取平均值。以吸附平衡时混合液上清igg平衡浓度为横坐标,对应的单位体积igg结合量为纵坐标,绘图并根据langmuir等温吸附方程计算静态饱和吸附容量。
按照相同方法测定igg溶液ph分别为6.0、7.4、8.0和8.9时功能化交联琼脂糖凝胶对igg的静态饱和吸附容量。langmuir等温吸附方程为:
qe=qm×ce/(kd+ce)
其中,qe(mg/g)和ce(mg/ml)为吸附平衡时的吸附量和浓度;qm是吸附剂的饱和吸附容量;kd是跟抗体与吸附剂结合强度有关的常数。
上述实验结果见附图3;由附图3结果表明,在溶液ph=7.4时,苯并三唑功能化交联琼脂糖凝胶对igg的静态结合容量最大,达到140mg/ml;当igg溶液的ph偏离7.4时,igg的静态结合容量明显下降。因此在溶液ph=7.4时,苯并三唑功能化交联琼脂糖凝胶对抗体的吸附效果最好。
实施例7动态结合容量评价
采用实施例1-5所得到的功能化交联琼脂糖凝胶作为层析介质,选取内径为0.66cm的玻璃层析柱,连入到配套有蠕动泵和紫外检测器的层析系统中,进行测定。紫外检测器在280nm波长下检测信号。分别取0.5g上述抽至无水析出的功能化交联琼脂糖凝胶作为层析介质装柱,用2.0mg/ml的人igg上柱,上样流速为1.0ml/min,10%流穿后停止上样。平衡缓冲液洗出柱中残留的未结合抗体。通过检测上样前后igg总量的差值确定功能化交联琼脂糖凝胶层析介质的吸附量。
作为对照,以实施例5得到的6-氨基己酸为间隔臂经甲酸封闭的交联琼脂糖凝胶,重复上述实验,测定苯并三唑基团被氢原子替换后对抗体的动态结合容量。
上述实验结果见表1;由表1结果表明,在各个配基密度范围下,经5-羧基苯并三唑或5-氨基苯并三唑功能化的交联琼脂糖凝胶层析介质对人抗体都具有吸附能力,且对抗体的动态结合容量随配基密度和间隔臂的长度增加而增加;当配基密度为164μmol/ml时,经5-羧基苯并三唑功能化的交联琼脂糖凝胶对抗体的动态结合容量高达57.7mg/ml;吸附结果表明,在高密度下,功能化交联琼脂糖凝胶对抗体具有显著地吸附效果,且达到同类商品化mep吸附剂对抗体的动态吸附量的1.6倍以上。
此外,在各个几乎相同的配基密度下,经5-氨基苯并三唑功能化的交联琼脂糖凝胶对抗体的动态吸附容量与以1,6-己二胺为间隔臂的5-羧基苯并三唑功能化交联琼脂糖凝胶的动态吸附量相当。在配基密度为160μmol/ml时,两种功能化交联琼脂糖凝胶对抗体的动态接和容量几乎相等。这一结果表明,在抗体动态吸附过程中,与抗体相互作用并将其吸附到功能化交联琼脂糖凝胶表面的是苯并三唑基团,与苯并三唑5位上的取代基无关。
最后,对照实验结果表明,当苯并三唑被氢原子替换后,即使在配基密度为160μmol/ml时,经甲酸封闭的功能化交联琼脂糖凝胶对抗体的动态吸附容量最高仅为0.53mg/ml干胶。这一结果表明,只有经苯并三唑功能化后,交联的琼脂糖凝胶才能有效地结合抗体。
综上所述,苯并三唑基团在抗体抗体吸附过程中发挥至关重要的作用,且与苯并三唑5位上的取代基无关。
表1功能化交联琼脂糖凝胶对抗体的动态结合容量
实施例8人血浆抗体吸附选择性评价
采用实施例1中所得的配基密度为164μmol/ml干胶的功能化交联琼脂糖凝胶层析介质,选取内径为0.66cm的玻璃层析柱,连入到配套有蠕动泵和紫外检测器的层析系统中。紫外检测器在280nm波长下检测信号。分别取0.5g上述抽至无水析出的功能化交联琼脂糖凝胶装柱,用2.0ml新鲜人血浆上样(血浆ph在7.4),当紫外信号迅速上升时,收集流穿液并测定蛋白浓度。上样平衡后,用ph7.4的缓冲液平衡柱子,除去未结合的蛋白。重复上述实验2次,第一次用ph4.0的柠檬酸洗脱液(0.01mol/l)洗脱结合的蛋白,第二次使用ph3.0的柠檬酸洗脱液(0.01mol/l)洗脱结合的蛋白,收集洗脱液并测定蛋白浓度。对收集的蛋白溶液及原液用聚丙烯酰胺凝胶电泳(sds-page)分析不同ph条件下洗脱得到的蛋白组成,使用灰度扫描软件计算洗脱液中抗体纯度并计算抗体的回收率。
上述实验结果见附图4;由附图4结果表明,与人血浆原液相比,经功能化交联琼脂糖凝胶吸附后的人血浆,其抗体含量明显降低,且洗脱的蛋白(即结合到功能化交联琼脂糖凝胶)主要成份为人抗体。这一结果表明该功能化的层析介质在复杂的蛋白溶液中具有特异性吸附人抗体的能力,具有在抗体分离工业及自身性免疫疾病的血液净化治疗潜在的应用。
表2为经灰度扫描后不同ph洗脱下抗体的纯度及回收率。由表2结果表明,在ph4.0的柠檬酸洗脱液洗脱下,功能化交联琼脂糖凝胶从人血浆中分离的抗体纯度超过95%,回收率达到72%以上;当降低洗脱液的ph,会引起洗脱液中杂蛋白的增加,但可以有效提高抗体回收率,抗体的回收率可以达到84%。因此,可根据分离抗体的目标不同,选择合适的洗脱ph。例如,在工业抗体分离方面为得到高纯度的抗体,可使用较高ph的洗脱液进行洗脱;而在血液净化方面对回收的抗体的纯度要求较低,因此可使用较低ph洗脱结合的抗体,以降低再生成本。
表2不同ph洗脱下抗体的纯度及回收率
自身免疫性疾病是机体自身产生的抗体破坏、损伤自身的组织和细胞成分,导致组织损害和器官功能障碍的原发性免疫性疾病。通过去除机体内多余的抗体可有效地治疗自身免疫性疾病,而实施例8实验结果表明,功能化凝胶能够从人血浆中特异性的识别并吸附抗体,因此可用于去除机体内多余的抗体成分,具有作为在自身免疫性疾病的血液净化治疗中应用的潜力。
同样地,实施例8实验结果表明,即使在抗体浓度含量较低的蛋白溶液中,苯并三唑吸附剂也具有特异性识别抗体的能力,说明它能够应用于抗体浓度更高的动物细胞培养上清中,以分离纯化抗体,从而具有作为新型疏水电荷诱导配基应用于抗体工业化生产的潜力。
实施例9测定离子强度对抗体吸附的影响
采用实施例1所得到的高配基密度的功能化交联琼脂糖凝胶作为层析介质,选取内径为0.66cm的玻璃层析柱,连入到配套有蠕动泵和紫外检测器的层析系统中,进行测定。紫外检测器在280nm波长下检测信号。取0.5g上述抽至无水析出的功能化交联琼脂糖凝胶作为层析介质装柱,用含氯化钠浓度为0.15mol/l的人igg溶液(2.0mg/ml)上柱,上样流速为1.0ml/min,10%流穿后停止上样。平衡缓冲液洗出柱中未结合蛋白。通过检测上样前后igg总量的差值确定功能化交联琼脂糖凝胶层析介质的吸附量。按照同样的方法,测定氯化钠浓度为0.5和1.0mol/l的人igg溶液上样后功能化交联琼脂糖凝胶层析介质对抗体的吸附量。作为对照,取不含氯化钠的人igg溶液(2.0mg/ml)上样,测定上样后功能化交联琼脂糖凝胶层析介质对抗体的吸附量。
上述实验结果见附图5;由附图5可知,在所测定的离子强度范围内,功能化交联琼脂糖凝胶层析介质动对抗体的动态结合容量没有明显下降,说明层析介质对抗体的吸附过程受盐离子的影响较小。因此,在抗体分离时,无需对料液的离子强度预处理即可直接上样,可有效地节约抗体的分离成本。
实施例10测定苛性条件下层析介质的理化稳定性
采用实施例1所得到的功能化交联琼脂糖凝胶作为层析介质,分别在75%乙醇、0.1mol/l氢氧化钠和盐酸溶液中震荡处理24h,另取一份进行高温灭菌。作为对照,取功能化交联琼脂糖凝胶置于1.0mol/l的氯化钠溶液中震荡处理24h。处理结束后,使用去离子水多次冲洗功能化交联琼脂糖凝胶以除去处理液。按照实施例5方法测定不同处理下功能化交联琼脂糖凝胶对人igg的动态吸附量。
上述实验结果见附图6;由附图6结果表明,功能化交联琼脂糖凝胶层析介质经过上述条件处理后,对抗体的动态结合容量未发生明显的改变,说明所得到的功能化交联琼脂糖凝胶作为层析介质在强碱、强酸中浸泡,及经过高温灭菌后,依旧保持很好的稳定性。
- 还没有人留言评论。精彩留言会获得点赞!