用于使用网络失调鉴定药物作用机制的方法和系统与流程
本申请要求于2014年5月9日提交的美国临时申请系列号61/991,005和于2014年7月21日提交的美国临时申请系列号62/027,045的优先权,这些申请通过引用整体并入本申请。
关于联邦资助的研究的声明
本发明是借助政府支持在由美国国立卫生研究院(NIH)授予的资助号U01 CA 164184、U01 HL 111566和U54 CA 121852资助下完成的。政府对本发明具有一定的权利。
发明背景
化合物的作用机制(MoA)可被定义为化合物籍以产生其药理作用的生物化学相互作用物和效应物的集合,所述作用机制通常是细胞环境特异性的。MoA可用于新药的开发,了解新药的副作用,以及药物重新定位。然而,这样的鉴定可能具有挑战性,是昂贵的,并且需要大的实验设置。
通过某些实验和计算策略仅部分解决这些挑战。许多实验方法依赖于直接结合测定,诸如亲和纯化或亲和层析测定。这些方法通常可被局限于鉴定高亲和力结合靶标,而不是负责化合物在组织中活性的全蛋白库。因此,这些方法可能错过某些间接效应物,以及可具有期望的药理学性质或驱动不期望的副作用的低亲和力结合靶标。例如,可以针对所有激酶筛选蛋白激酶抑制剂,但是可错过其他相关靶标,如通过MET酪氨酸受体激酶抑制剂tivantinib作为微管抑制剂的重新分类所显示的。另外,某些方法仅适合于体外研究,并且可错过由旁分泌、内分泌体内和接触信号产生的以及特定组织环境中的复杂效应。
虽然也已开发了化学信息学方法,但是某些技术通过利用结构和基因组信息的整合、文本挖掘算法或用于数据挖掘的机器学习方法来评估MoA相似性或特异性小分子/靶标相互作用。因此,它们可以依赖于药物分子和靶蛋白质二者的详细三维结构或者依赖于相关MoA化合物的现有知识(来源于文献或数据库)。
还已经结合用于MoA分析的计算方法开发了基于在化合物于细胞系中的扰动后系统基因表达谱(GEP)的技术。这些技术的范围可以为基于差异表达分析以比较未知MoA的新化合物的简单功能基因表征到扰动GEP的大参考汇编。后者可使用多种相似性度量来评估代表疾病相关细胞系对具有确定的MoA的化合物的扰动或RNAi介导的基因沉默测定的响应的GEP的相似性。然而,某些方法本质上大多是比较性的,因此不太适合MoA的从头阐明或识别微妙的MoA差异,从而导致例如不希望的毒性。
另一个选择是基于网络的方法,所述方法不是集中于个体基因的特征,而是通过整合其相互作用伴侣或途径的表达来估算基因产物活性的变化。这可以允许使用环境特异性知识和分子相互作用数据,同时还通过整合多个基因的信号来提供稳健性。然而,某些方法或者依赖于介导化合物活性的途径的事先知识,使得它们不适合于全基因组分析,或者需要非常大的样本大小(n>100),因此使得它们即使对于小的化合物文库也是不切实际的。
因此,需要开发用于鉴定化合物作用机制的改进方法。
发明概述
本发明公开的主题提供了用于鉴定化合物的作用机制的方法和系统。
根据公开的主题的一个方面,提供了用于使用网络失调来鉴定化合物的作用机制的方法。在示例性实施方案中,所述方法可包括选择涉及至少第一基因的至少第一相互作用,并且例如使用处理装置测定第一基因和处于对照状态中的一个或多个基因的基因表达水平的第一n维概率密度。在一些实施方案中,n可以等于2;一个或多个基因可以是一个基因。所述方法还可以包括测定在使用至少一种化合物处理后第一基因和一个或多个基因的基因表达水平的第二n维概率密度,并估算第一概率密度与第二概率密度之间的变化。在一些实施方案中,估算可以包括使用Kullback-Leibler散度。
所述方法还可包括确定所估算的变化是否是统计上显著的。在一些实施方案中,该特征可包括通过从随机基因对(无论基因是否共享网络边缘)估算的105个Kullback-Leibler散度值产生的零分布,提供网络中每个边缘失调的P值。在一些实施方案中,估算包括基于一个相互作用接着一个相互作用的估算。
在一些实施方案中,选择第一相互作用可以包括选择m个相互作用,以及针对m个相互作用中的每一个重复该过程。例如,m个相互作用可以是在调控网络中以第一基因结束的每个相互作用。所述方法可包括如果所估算的变化是统计上显著的,则确定每个相互作用是否失调。所述方法可包括至少部分地基于每个相互作用的显著性来确定第一基因是否失调。在一些实施方案中,确定第一基因是否失调可以至少部分地基于对每个相互作用的显著性进行积分。在一些实施方案中,对显著性进行积分可包括估算对第一基因的线性拟合的残差,和使用所得残差的协方差矩阵作为对Brown方法的输入。
第一基因可以包括多个基因,并且所述方法可以包括针对多个基因重复该过程。在一些实施方案中,所述方法可包括通过选择被确定为显著的基因来鉴定化合物的作用机制。化合物可包括多种化合物,并且所述方法可包括针对多种化合物中的每一种进行重复处理,以及鉴定具有类似药理学作用的两种或更多种化合物。
在公开主题的另一个示例性实施方案中,提供了鉴定具有类似药理作用的化合物的方法。示例性方法可包括选择涉及至少第一基因的第一相互作用。所述方法还可包括例如使用处理装置测定第一基因和处于对照状态中的一个或多个基因的基因表达水平的第一n维概率密度。在一些实施方案中,n可以等于2;一个或多个基因可以是一个基因。
所述方法可包括测定在使用第一化合物的第一化合物处理之后第一基因和一个或多个基因的基因表达水平的第二n维概率密度,并且估算第一概率密度与第二概率密度之间的变化。所述方法还可包括确定所估算的变化是否是统计上显著的,以及如果所估算的变化是统计上显著的,则确定相互作用是否失调的。
可针对m个相互作用中的每次重复所述方法,其中m个相互作用包括在调控网络中以第一基因结束的每个相互作用。所述方法可包括至少部分地基于每个相互作用的显著性来确定第一基因是否失调。在一些实施方案中,确定第一基因是否失调可以至少部分基于对每个相互作用的显著性进行积分。在一些实施方案中,对显著性进行积分可包括估算对第一基因的线性拟合的残差,并使用所得残差的协方差矩阵作为对Brown方法的输入。
可以针对多个基因和通过选择失调的基因鉴定第一化合物的作用机制重复所述方法。对于使用多种化合物的多种化合物处理,也可以重复所述方法。另外,所述方法可包括鉴定具有类似药理作用的两种或更多种化合物。在一些实施方案中,鉴定具有类似药理作用的两种或更多种化合物可包括至少部分地基于它们的预测的作用机制之间的相似性的显著性来预测两种或更多种化合物享有相似的药理作用。在一些实施方案中,估算可包括使用Kullback-Leibler散度。在一些实施方案中,估算包括基于一个相互作用接着一个相互作用的估算。
本文的描述仅示出所公开的主题的原理。鉴于本文的教导,对所描述的实施方案的各种修改和改变对于本领域技术人员将是显而易见的。因此,本文的公开内容旨在说明而非限制所公开的主题的范围。
附图简述
图1举例说明使用网络失调来鉴定化合物的作用机制的方法。
图2举例说明鉴定具有类似药理作用的化合物的方法。
图3举例说明根据所公开主题的示例性算法的示意图。
图4举例说明所公开的实施方案与t检验之间的比较和生物表现预测。
图5举例说明DP14数据集的示例结果。
图6举例说明预测由长春花新碱和丝裂霉素C产生的失调的实验验证。
图7举例说明鉴定运行所公开的实施方案的最低要求。
图8举例说明来自GEO数据集的示例结果。
图9举例说明检测化合物相似性的示例性预测。
图10举例说明化合物相似性的性能分析。
图11举例说明所公开的实施方案鉴定六甲蜜胺的MoA。
图12举例说明估算相互作用与p值的Brown方法校正之间的相关性。
发明详述
本文提供的方法和系统可用于鉴定化合物的作用机制和鉴定具有类似药理作用的化合物。所公开的主题可通过使用调控网络评估化合物扰动之后它们的分子相互作用库的所有组分的全局失调来鉴定相关的MoA蛋白。将结合本文中称为通过网络失调检测作用机制(下文中称为“DeMAND”)的示例方法来解释所公开的主题,以通过使用小型基因表达谱(GEP)数据集(例如,n≥6个样品)来询问组织特异性调控网络来阐明化合物MoA,所述数据集代表体外或体内化合物特异性扰动。
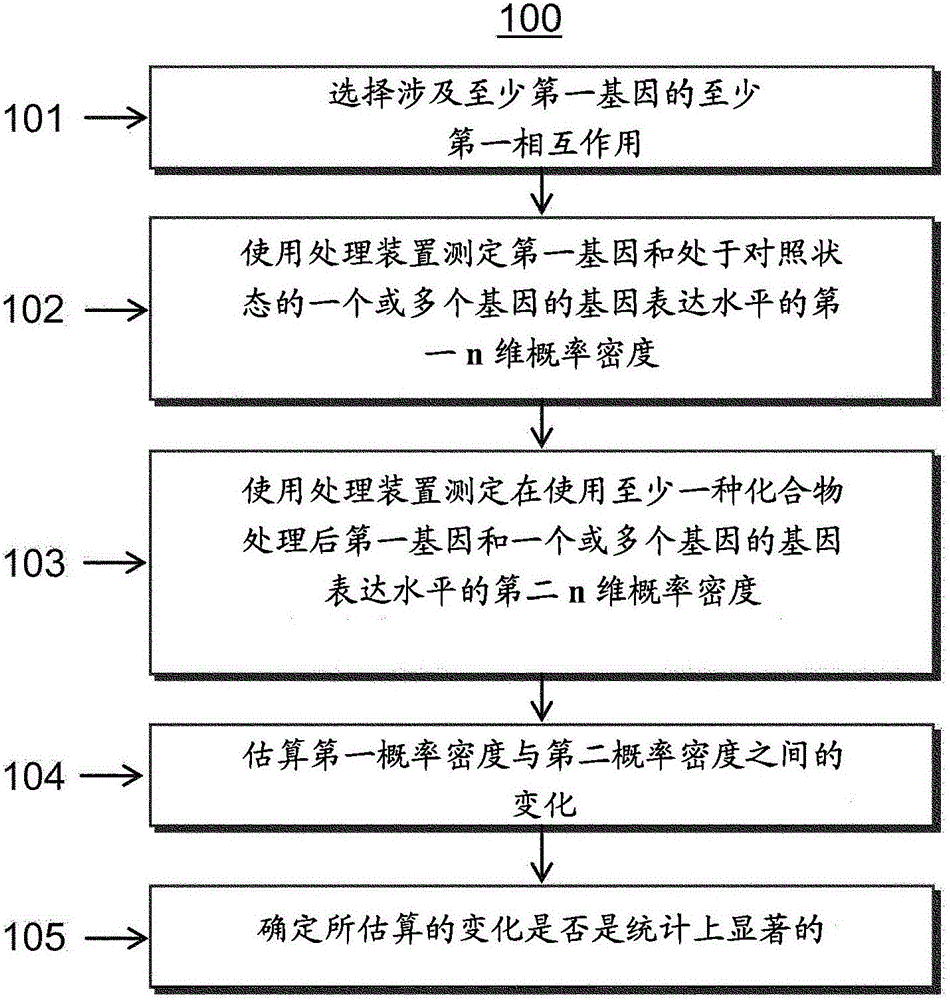
为了说明而非限制的目的,图1显示了用于使用网络失调来鉴定化合物的作用机制的方法(100)。所述方法可包括选择涉及至少第一基因的至少第一相互作用(101)。在(102)处,所述方法包括使用处理装置测定第一基因和处于对照状态中的一个或多个基因的基因表达水平的第一n维概率密度。在一些实施方案中,n可以是2,并且一个或多个基因可以是一个基因。在(103)处,所述方法包括使用处理装置测定在使用至少一种化合物处理后第一基因和一个或多个基因的基因表达水平的第二n维概率密度。在(104)处,所述方法可包括估算第一概率密度与第二概率密度之间的变化。在一些实施方案中,估算可以基于一个相互作用接着一个相互作用进行估算。估算可使用Kullback-Leibler散度(在下面更详细地描述)。在(105)处,所述方法可包括确定所估算的变化是否是统计上显著的。例如,可如下更详细地描述的那样确定所估算的变化是否是统计上显著的。在一些实施方案中,所述方法可包括使用通过从随机基因对估算的105个Kullback-Leibler值产生的零分布,和提供网络中每个边缘失调的P值。
为了说明而非限制的目的,图2显示了鉴定具有类似药理作用的化合物的方法。在(201)处,所述方法可包括选择涉及至少第一基因的至少第一相互作用。在(202)处,所述方法可包括使用处理装置测定第一基因和处于对照状态中的一个或多个基因的基因表达水平的第一n维概率密度。在(203)处,所述方法可包括使用处理装置测定在使用至少一种化合物处理后第一基因和一个或多个基因的基因表达水平的第二n维概率密度。在(204)处,所述方法可包括估算第一概率密度与第二概率密度之间的变化。例如,估算可以基于一个相互作用接着一个相互作用进行估算,并且可以使用Kullback-Leibler散度。在(205)处,所述方法可包括确定估算的变化是否是统计上显著的。在(206)处,所述方法可包括如果所估算的变化是统计上显著的,则确定相互作用是否失调。在(207)处,可针对m个相互作用中的每一个重复所述方法。m个相互作用可包括在调控网络中以第一基因结束的每个相互作用。在(208)处,所述方法可包括至少部分地基于每个相互作用的显著性来确定第一基因是否失调。在(209)处,可针对多个基因重复所述方法。在(210)处,所述方法可包括通过选择失调的基因来鉴定第一化合物的作用机制。在(211)处,可针对使用多种化合物的多种化合物处理重复所述方法。在(212)处,所述方法可包括鉴定具有类似药理作用的两种或更多种化合物。
实施例:DeMAND
给定包括转录、信号传导和蛋白质-复合物相关相互作用的候选基因产物G(即,其调节子)的潜在相互作用,如果G属于化合物的MoA,则可假定化合物的活性将使其调节子失调。因此,可通过测量成对调节子相互作用之间的基因表达的联合概率密度p(G,G')的变化来评估此类失调。这可通过直接或间接调控机制(例如,反馈回路)捕获化合物对候选MoA基因表达和相互作用伴侣表达的调节的直接效应。
例如,考虑调控一组转录靶标的候选MoA基因产物G。靶向抑制剂将显著改变G及其靶标的联合表达概率密度,因为后者的表达将受到影响,而G的表达通常不受影响。因此,在化合物扰动之后,可以观察到相互作用概率密度p(G,Gi)的显著变化,如图3中和下面的实验程序讨论中的3个失调相互作用的概率密度所示。
Kullback-Leibler散度(KLD)可提供确定的且有效的度量来定量评估一个或多个变量的概率密度的变化。当一个概率密度用作另一个概率密度的近似时,KLD可评估信息的损失,因此可在信息理论的背景中进行解释。因此,对于每个候选失调相互作用可在化合物扰动之前和之后估算在其调节子中每个G'基因的概率密度p(G,G')的KLD的统计显著性。然后可跨调节子相互作用整合单个KLD分析的统计显著性,产生化合物介导的G失调的全局统计评估。为了避免因相互作用依赖性而高估这样的整合显著性,可使用补偿相关证据的整合的改良Brown方法。然后基于候选MoA基因的全局统计数据对其进行分级(rank)。
为了鉴定每种目标基因产物的调节子,可使用下面描述的一组确定的网络逆向工程算法。然而,本发明所公开的主题不需要这样的算法,而是可以使用由替代技术(计算和实验技术)产生的网络。
评价了在针对14种选定的化合物的扰动数据集(DP14)中DeMAND推断的MoA基因的准确度。这包括在以两个浓度和三个时间点一式三份地利用14种不同化合物(其中11种已建立主要靶标,参见实验程序)并以DMSO作为对照介质进行扰动之后弥漫性大B细胞淋巴瘤细胞(OCI-LY3)的276个GEP。使用代表正常和肿瘤相关的人B细胞二者的226个U133p2 GEP的数据集产生用于这些分析的网络(参见实验程序)。尽管DeMAND可预测化合物靶标(即,高亲和力结合蛋白)和效应物/调节剂二者,但它的性能被证明是系统地以前者为基准的。
在该分析中,DeMAND将11种测试化合物中7种的主要靶标鉴定为统计上显著的(其假发现率FDR≤0.1)(见下文和图4A)。由于在该分析中使用的GEP在多个时间点(6、12和24小时)获得,因此评估了在个体时间点的分析是否可以比跨多个(例如,所有)时间点的整合更具信息性。仅在这些时间点中的某些时间点可以高度特异性地预测几个已确定的靶标(图4B),这与化合物活性可在不同时间尺度上被介导的预期相一致。又,除2种化合物(monastrol和多柔比星)外的所有化合物在多个(例如,所有)时间点上的整合分析表现与其个体时间点的分析一样好或者更好。对于这些,仅当使用在特定时间点的GEP时,直接靶才是显著的。总的来说,11种化合物中9种化合物的主要靶标是从多点分析或从单点分析阐明的。
将DeMAND在主要靶标推断上的性能与差异表达分析(通过t检验统计)的性能相比较。除blebbistatin外,DeMAND的预测均系统地超越t检验分析;对于blebbistatin,两种方法都未将主要靶标(肌球蛋白II)鉴定为统计上显著的(图4A)。当如图5A所示比较通过两种方法在其最高预测(灵敏度)中所鉴定的直接靶标的分数时,在排在前面的100个预测当中,在预测已确定的化合物靶标中DeMAND相较于t检验分析其灵敏度几乎是5倍(15%对3%),这是高度统计上显著的(分别为p=5x10-4,和p=0.06,通过χ2检验)。此外,通过差异表达分析鉴定的显著性靶标通过DeMAND分析也是显著的,但不是相反的。总体上,考虑到接受者操作特征(ROC)曲线下完整面积(AUC),所述面积是被设计用于获得方法在预测直接靶标中的灵敏度与特异性之间的折衷的阈值自由度量,DeMAND始终优于t检验,分别是AUC=0.70(p值≤2×10-16,通过每种化合物的个体Mann-Whitney p值的费希尔积分)对AUC=0.60(p值=3.5x10-7),反映其显著更高的灵敏度和特异性(图4C)。
为了进一步评估DeMAND对不是高亲和性化合物靶标的MoA蛋白质的性能,进一步分析集中于四种化合物中的两种:喜树碱和多柔比星,它们的直接靶标被分析错过了。尽管缺少它们的主要靶标,但DeMAND有效地鉴定了这两种化合物的关键MoA蛋白。具体来说,喜树碱(TOP1抑制剂)和多柔比星(TOP2A抑制剂)严重破坏DNA修复和有丝分裂。DeMAND鉴定了这些化合物的最有统计学意义的基因中的作为TOP1和TOP2A抑制的下游效应物的GADD45A、CDKN1A、PCNA、AURKA、PLK1和CCNB1(图5B),对于两种化合物它们的大多数在前20个中。
更具体地,GADD45A(生长阻滞和DNA损伤诱导型基因45A)(一种确定的DAN损伤反应的效应物)通过与CDKN1A(细胞周期蛋白依赖性激酶抑制剂1A)和PCNA(增殖细胞核抗原)形成蛋白质复合物(一种高保真DNA复制和切除修复所需的DNA聚合酶δ的持续性因子)起作用。进而,如果检测到DNA损伤,则CDKN1A、PCNA和GADD45A在RNA或蛋白质水平上调控CCNB1(细胞周期蛋白B1,G2/M细胞周期检查点的关键效应物)、PLK1(polo样激酶1)和AURKA(Aurora激酶A,有丝分裂调节剂)的活性。在这6个基因中,在这些化合物扰动后,只有GADD45A和CDKN1A差异表达,虽然程度低得多。因此,DeMAND鉴定出通过差异表达检测不到或分级较低的关键MoA蛋白。
经DeMAND推断的蛋白质的详细评估成功地突出了在具有在基因表达水平不可检测的类似主要靶标的化合物的MoA中的关键差异和共性。例如,喜树碱(TOP1)、多柔比星(TOP2A)和依托泊苷(TOP2A)是拓扑异构酶(TOP)抑制剂,导致TOP-DNA可切割复合物的共价捕获和单链或双链断裂的诱导。一致地,DeMAND在其推断的MoA中鉴定了共同的足迹,如上所示。然而,它还鉴定了多柔比星的特异性效应物,例如KAT5/TIP60(分级为第4位),表明与另外两种TOP抑制剂的潜在相关差异,如图5B所示。事实上,与依托泊苷和喜树碱相反,多柔比星也是一种强DNA嵌入剂,其在大规模KAT5依赖性组蛋白乙酰化后,诱导组蛋白从开放染色体位点释放(组蛋白回收),导致细胞周期阻滞。再次,差异表达没有鉴定出(甚至在前1000个基因中)多柔比星的潜在效应物诸如KAT5和PCNA。该算法还将SIK1鉴定为多柔比星的特定效应物(分级为第36位)。SIK1用于维持心脏祖细胞(CPC),从而精确定位化合物的最突出的治疗限制性不良事件,即伴有充血性心力衰竭的心肌病。
DeMAND还可基于它们共同的预测的MoA基因对化合物成功分层,强调它在预测化合物的MoA中的特异性。例如,14种测试化合物中的5种化合物是DNA损伤剂(即,喜树碱、多柔比星、依托泊苷、丝裂霉素C和长春花新碱)。DeMAND预测所有这些化合物的最具统计学显著的MoA推断基因中的GADD45A(规范的DNA损伤诱导型基因),以及其众所周知的相互作用基因,诸如CDKN1、CCNB1PCNA和AURKA[32-35](图5C)。又,相同的基因对于不诱导DNA损伤的化合物是不显著的(图5C),表明DeMAND可以推断高度特异性的化合物MoA蛋白。相反,差异表达表现差,无法将这些基因中的大多数鉴定为统计上显著的。因此,DeMAND可提供关键信息,以鉴定小分子化合物的“命中目标(on-target)”以及潜在有害的“偏离目标(off-target)”效应。
为了说明DeMAND对于阐明新型MoA蛋白是有效的,经DeMAND推断的MoA蛋白可以是针对长春花新碱(一种有丝分裂纺锤体中微管形成的抑制剂)和丝裂霉素C(一种抗肿瘤抗生素)。DeMAND在其前5个预测的基因中成功鉴定了长春花新碱(TUBB)的已知的高亲和力靶标以及CCNB1(一种已知的微管活性标志物)。前5个中的其他3个基因包括VHL、RPS3A和NFKBIA。尽管已知这些基因中的两个(RPS3A和VHL)影响人细胞系中的有丝分裂纺锤体组装,但是它们在介导/调节长春花新碱活性中的功能是未知的。
在沉默这些基因后,用抗微管蛋白抗体探测微管网络,证实了RPS3A(但不是VHL、CCNB1或NFKBIA)的沉默破坏了贴壁U-2-OS细胞中的微管(图6A)。因此,为了进一步验证这些基因在介导长春花新碱活性中的作用,在siRNA介导的每个基因的沉默之后,进行U-2-OS细胞中的剂量-反应曲线测定(参见实验程序)。这些测定证实所有这些基因,除了NFKBIA,是关键的长春花新碱活性效应物和介体。VHL沉默使长春花新碱灵敏度增加超过两倍(图3B),而RPS3A和CCNB1沉默具有相反的作用。因此,5个DeMAND推断的基因中的3个基因被实验验证为长春花新碱活性调节剂,而第四个(TUBB)代表其主要靶标。相反,这些基因没有一个显著差异表达,因此不能通过更常规的方法检测。这表明,对于一些化合物,算法的假阳性率可以低至20%。
DeMAND还推断JAK2激酶是唯一的丝裂霉素C MoA蛋白(即,通过DeMAND分析,JAK2对于其它化合物是不显著的)。这可能是重要的,因为JAK2的组成型活性可引起淋巴细胞的化学抗性,并且此外,最近的研究表明组成型JAK2活性可以控制DNA损伤、修复和重组事件的结果。因此,通过测量用不同量的充分表征的JAK2抑制剂TG101348处理后该化合物的剂量-反应曲线,测试JAK2抑制对丝裂霉素C活性的影响(图6C,参见实验程序)。该实验揭示了JAK2抑制与丝裂霉素C活性之间的显著的剂量依赖性拮抗作用,因此证实JAK2是丝裂霉素C活性的关键效应物。
最后,分析了DeMAND对抗肿瘤和免疫调节剂雷帕霉素进行推断的结果。虽然DeMAND不能预测最高亲和力靶标MTOR和FKBP1A,但MTOR途径下游的许多基因高度富集于排在前面的DeMAND推断的基因中(图4E,参见实验程序),包括许多核糖体基因。这不是多效性结果,因为对于核糖体基因具有显著富集的唯一其它化合物是放线酮,也已知放线酮抑制核糖体活性,因此再次突出DeMAND预测的特异性。
为了评估所述方法的广泛利用潜力,对其稳健性和其数据要求进行了基准测试。对作为网络准确度和大小以及扰动数据集大小的函数的DeMAND的性能进行了评估。首先,使用从254个Affymetrix U95av2 GEP的不同数据集重建的独立的B细胞基因调控网络比较所获得的结果,如下所述。对于该比较,使用U95av2网络,通过基因集富集分析(GSEA),针对使用U133p2网络推断的那些基因,测试了统计上显著的DeMAND推断的基因(FDR≤0.1)的富集。分析证实DeMAND的预测几乎相同,与网络模型无关(GSEA,p<1x10-9,图7A),证实DeMAND的性能在很大程度上独立于特定的分析平台和用于网络组装的数据集。进一步的分析表明,当至多60%的网络相互作用被随机去除时,DeMAND预测几乎不受影响(参见图7B)。类似地,当使用代表化合物扰动的6个或更多个样品时,对GEP的二次取样显示DeMAND预测实际上是相同的(图7C)。总之,这些数据表明,DeMAND对于网络噪声和尤其是假阴性相互作用是高度稳健的,并且其可以成功地应用于小至6个处理样品和6个未处理对照的数据集。
一旦客观地评估了对稳健算法性能的要求,就可选择在基因表达综合(GEO)数据库中所选择的13个额外数据集(GEO13),代表在化合物扰动后的细胞系GEP(表1)。这些数据集被限制于具有已确定的主要靶标的化合物,其中至少6种谱(对于化合物处理的和对照样品二者)是可用的,并且其中可以组装背景特异性调控网络。这包括7个人乳腺癌和6个人B细胞淋巴瘤扰动数据集。类似于PD14数据集,DeMAND将一个或多个确立的直接靶标鉴定为对于62%的这些化合物扰动(FDR≤0.1,图8A)是统计上显著的,同时显著优于基于t检验的方法(AUC分别为0.82对0.74,通过每种化合物的个体Mann-Whitney p值的费希尔积分,p值=2.2x10-16对p值=5.9x10-8)(图8B)。再次,这些差异在排在前面的预测的化合物中尤其相关,其中DeMAND再次实现比差异表达分析好大约5倍的性能(图8C)。
表1
表1.来自GEO数据库的13个化合物扰动数据集
在建立DeMAND在预测参照化合物的MoA基因中的性能时,检查预测其药理学相似性的不同化合物的推断的MoA重叠。首先,计算DP14化合物对的显著的经DeMAND推断的MoA的重叠的统计显著性(FDR≤0.1,通过费希尔精确检验)(参见图9A)。在91个潜在的化合物对中,6个最相似的化合物对仅包括拓扑异构酶抑制剂和其它DNA损伤剂(依托泊苷、多柔比星、喜树碱和丝裂霉素C)。因此,DeMAND成功评估了拓扑异构酶抑制剂与其他DNA损伤剂之间的化合物的MoA高度相似性,即使它不能在推断的MoA基因中鉴定出TOP1或TOP2A。因此,效应蛋白可具有与阐明化合物相似性中的直接靶标相同的信息性。
为了进一步评价该假设,将所述方法应用于更大的化合物扰动数据集(PD92),其代表3种不同B细胞淋巴瘤细胞系(OCI-LY3、OCI-LY7和U-2932)在用92种独特的FDA批准的后期实验的工具性化合物的扰动后的基因表达谱(见实验程序)。因为在该数据集中每个化合物和细胞系仅有3种GEP可用,所以该数据集仅用于基于预测的MoA评估化合物对相似性,而不用于预测主要化合物靶标(参见实验程序)。
通过使用以下3个独立的数据源比较DeMAND推断的相似性与客观相似性评估来客观地评价DeMAND性能:(a)共享确定的靶标的化合物;(b)根据解剖治疗化学分类系统(ATC)的共享治疗和化学特征的化合物和(c)由癌症靶标发现和发展(CTD2)联盟评估的具有相关药物反应谱的化合物(参见实验程序)。后一数据集概括了代表针对代表多种肿瘤类型的257种不同的细胞系表征的338个独特化合物的剂量反应曲线载体。基于3个证据数据集中的每一个,作为显著对的数量(精密度曲线,图9B)的函数,评估验证的相似对的分数(精密度)。经DeMAND推断的对在来自3个证据数据集的对中高度富集,如由个别的(即,对于共享相同ATC类别、共同确定的靶标和在CTD2数据集中的高剂量-反应载体相关性的对,GSEA的p值分别=2×10-8、1.4×10-5和9×10-4,图10A)以及当一起时的(GSEA p值=7.6×10-7)证据评估的。例如,经DeMAND推断的对相似性中前10个中的8个和前100个中的43个被3个数据集里的至少一个数据集验证了(通过费希尔精确检验,p值<2.2×10-16)。
DeMAND通过在各种精密度值一致地实现更高的灵敏度,优于使用通过重叠统计上显著的差异表达的基因获得的相似性(例如,通过t检验统计)(图10B)。DeMAND也胜过另一种方法MANTRA,所述MANTRA使用相互基因集富集分析来计算相似性,同样通过在各种期望的精密度值下实现更高的灵敏度(图10B)。值得注意的是,虽然MANTRA可以评估化合物MoA相似性,但其并非旨在阐明特定的MoA基因。因此,DeMAND优于MANTRA,即使其仅使用少数预测为化合物MoA的一部分的基因而不是全基因表达特征进行MoA相似性分析。这进一步表明由DeMAND推断的MoA在生物学和机械学上是相关的。
最后,评估了由所述方法预测的化合物对相似性与其基于CTD2的相似性之间的相关性。DeMAND预测实现了显著的斯皮尔曼相关(ρ=0.59,p值=7.8×10-5,图10C),而t检验和MANTRA方法二者均未获得统计上显著的相关性(图10D,10E)。因此,DeMAND可在单个细胞系中处理后,仅使用GEP预测具有相似药理作用和活性谱的化合物。
为了测试具有统计上显著的经DeMAND推断的MoA相似性的两种化合物是否可能具有共同的靶标和效应物,将六甲蜜胺和柳氮磺胺吡啶鉴定为其中至少一种化合物的MoA是未知的对中的具有最高的经DeMAND推断的MoA相似性(p值=9.91×10-81)的两种化合物。六甲蜜胺是FDA批准的抗肿瘤药物,没有已知的靶或效应物支持其药理作用。另一方面,柳氮磺吡啶可以抑制系统xc-,胱氨酸-谷氨酸反向转运蛋白,从而防止胱氨酸进入细胞质并还原为半胱氨酸。因为半胱氨酸是谷胱甘肽生物合成中必需的代谢物,所以柳氮磺吡啶耗尽细胞谷胱甘肽,从而使依赖还原型谷胱甘肽(GSH)作为辅因子的酶失活,包括谷胱甘肽过氧化物酶4(GPX4)。这导致脂质活性氧(ROS)的毒性积累。
基于推断的MoA相似性,测试了六甲蜜胺是否也可以调节系统xc--GPX4途径。U-2932细胞用六甲蜜胺处理,并且使用Ellman试剂评估其GSH水平(图11A)。柳氮磺吡啶用作U-2932细胞中GSH耗尽的阳性对照,证实化合物处理后GSH水平的耗尽。相反,六甲蜜胺即使在24小时浓度下其IC50加倍后也不耗尽GSH水平,这表明该化合物在该途径中可以靶向GSH下游的机制。因此,U-2932细胞用六甲蜜胺处理,并制备细胞裂解物用于基于LC-MS的GPX4测定。将磷酸胆碱过氧化物(PC-OOH)(GPX4的特异性底物)添加至细胞裂解物中,通过[PC-OOH+H+]离子(m/z=790.5)的质量色谱图评估PC-OOH至PC-OH的还原。如图11B所示,未处理细胞的裂解物完全降低PC-OOH水平,不留下[PC-OOH+H+]离子(m/z=790.5)的残留信号。形成鲜明对比的是,来自六甲蜜胺处理的细胞的裂解物显示显著的[PC-OOH+H+]信号,表明PC-OOH还原的消除由GPX4抑制介导(实验程序)。实际上,由于GPX4是唯一已知的能够还原脂质氢过氧化物的酶,因此GPX4抑制是增加脂质-ROS水平所必需的。如所预期的,如通过BODIPY-C11染色和流式细胞术所评估的,柳氮磺吡啶和六甲蜜胺二者均被证实在U-2932细胞中诱导脂质-ROS积累(参见图11C和实验程序)。因此,DeMAND正确预测了两种以前不相关的药物柳氮磺胺吡啶与六甲蜜胺之间出乎意料的和引人注目的MoA相似性(参见图11D)。此外,这些结果表明六甲蜜胺为GPX4活性的新的抑制剂,并表明六甲蜜胺在患者中的抗肿瘤活性可能部分由于其酶活性的抑制。
DeMAND通过仅使用基因表达数据,基于全基因组范围评估化合物介导的蛋白调节子的失调来阐明化合物MoA。DeMAND可以可靠地鉴定MoA相关蛋白,后者可有效地用于评估任意化合物对的MoA和总体药理效应相似性。事实上,通过使用这种方法,人们可鉴定和实验验证以前未知的参与长春花新碱、丝裂霉素C和六甲蜜胺的MoA的几个基因。对于六甲蜜胺,人们还可鉴定和验证药物的新的药理作用(导致细胞死亡的增强的反应性氧化应激)。
DeMAND的稳健性分析证实,其预测在基因表达和网络变异性方面是稳健的,在至多60%的网络相互作用被去除时仍保持几乎不变。这可表明对网络中的假阴性相互作用的弹性。最后,与先前的方法不同,DeMAND可以可靠地用于非常小的扰动GEP集合(即,包含少至6个对照和6个扰动样品)。这可允许应用所述方法来阐明相对大的化合物组的MoA,例如以显示潜在的毒性偏离目标以及新型命中目标效应物和活性调节剂。这可支持将所述方法应用于若干由效应物产生的大规模库诸如基于集成网络的蜂窝签名的库(LINCS)数据集(代表在约4000种化合物扰动后的GEP)。另一个有利方面是算法的背景特异性性质,其允许探索特定的目标细胞环境(包括体内)中的化合物活性。
DeMAND利用在多个时间点和多种化合物浓度下获得的GEP的整合,从而简化实验设计,特别是当可以揭示MoA的精确浓度或时间点未知时。实际上,在没有具体知识的情况下,通过在多个时间点整合化合物反应,除了两种所测试的化合物之外,最佳地鉴定化合物靶标。又,当可获得时,对特定化合物活性时间尺度的了解也是有帮助的,如通过整合分析不能阐明的两种药物的直接靶标的鉴定所示(图4B)。总体上,如果化合物的时间依赖性响应是未知的,则可以选择多点分析。然而,多点和单点分析的比较可用于提供对化合物MoA的进一步了解。例如,与多点结果最相似的时间点的鉴定可以阐明化合物活性时间尺度。类似地,在两个连续时间点的分析的强一致性可表明可被整合分析所错过的MoA蛋白。本文所述的测定法已使用亚致死化合物浓度来避免利用与下游细胞死亡机制相关的机制污染化合物的MoA。
DeMAND预测是高度特异性的,允许将化合物分类为功能类似的组和鉴定与化合物MoA相关的途径。例如,对于DNA损伤化合物(喜树碱、多柔比星、依托泊苷、长春花新碱和丝裂霉素C),DeMAND正确预测了几个参与DNA损伤诱导反应的标志基因。特异性由以下事实证明:相关的MoA蛋白被推断用于DNA损伤诱导化合物,而不是其它化合物(包括表现出显著的多药理学的化合物,如H-7二盐酸盐或放线酮)。
在其他实施例中,可以显示多柔比星的高化合物-MoA特异性,其中DeMAND将KAT5(与最近的多柔比星特异性KAT5介导的组蛋白回收的发现一致)以及SIK1(心脏祖细胞(CPC)维持所需的基因)鉴定为关键的MoA-蛋白,提供了多柔比星与其已知的心脏毒性之间的潜在机械联系。在其他DNA损伤剂的MoA中也检测到SIK1(虽然分级/显著性比多柔比星低得多的),表明也应当被监测这些化合物的心脏毒性。在组合中,这些发现证实DeMAND不仅在预测直接化合物靶标方面有效,而且在预测关键的间接效应物蛋白方面也有效,因此允许MoA推断和鉴定可帮助阐明命中目标的药理学和偏离目标的毒性的潜在效应物。总体而言,DeMAND成功地鉴定了超过70%的测试化合物的直接靶标和间接MoA蛋白。尽管长春花新碱特异性新型MoA蛋白的实验验证表明实际的假发现率(FDR)可以低至20%,但是系统的FDR估算可能是困难的,因为化合物MoA被阐明得非常不足,导致显著的FDR高估。例如,在实验验证之前,发现长春花新碱推断的MoA蛋白的FDR为80%,只有TUBB是已确立的化合物靶/效应物。然而,前5个推断的MoA蛋白的系统验证显示FDR不超过20%。
DeMAND依赖于高质量背景特异性基因调控网络的存在,所述网络可代表对特定细胞背景的限制。然而,考虑到诸如癌症基因组图谱(TCGA)和其他相关联盟的大规模项目产生的丰富的数据,以及用于背景特异性网络逆向工程的越来越准确和全面的方法的可用性,这种限制至多是临时的。然而,细胞背景特异性调控网络的可用性不能保证在网络中被很少表示的MoA蛋白的鉴定。这可能发生,例如,因为个体基因的表达可以通过特定平台或由于通过逆向工程方法引入的假阳性而被很差地评估。例如,对于blebbistatin(肌球蛋白II抑制剂),通过使用U95av2网络,DeMAND鉴定了PTK2B、GRB2和FYN,其均为肌球蛋白II磷酸化的直接调节剂和肌球蛋白II扰动的响应者(参见图4D)。然而,由于在U133p2网络中缺乏GRB2表示,当使用U133p2网络时,不能推断该基因。同样重要的是,强调使用来自STRING数据库的高质量无关背景的网络对DP14和DP92数据集的DeMAND分析仍然能够鉴定遍在靶和效应物(例如,参与细胞周期和DNA损伤修复机制的那些靶和效应物),但在化合物相似性分析和具有背景特异性功能/表达的基因的鉴定中表现出较低的性能。这表明非背景特异性网络仍然可以用于DeMAND分析,虽然假阳性和阴性预测增加。这可能包括代表化合物特异性差异和潜在毒性相关效应物的基因,其只有在使用背景特异性网络时才被最佳地突显。
实验程序
DP14数据集:该数据集含有用14种不同的个体化合物处理的OCI-LY3细胞系(人弥漫性大B细胞淋巴瘤细胞系)的GEP,并且在化合物处理后6hrs、12hrs和24hrs进行分析(全部一式三份)。对于处理,使用两种不同浓度的化合物,对应于在24hrs时的IC20和在48hrs时的IC20。将在3个不同时间点以一式八份分析的DMSO处理的样品的GEP作为对照,得到来自该数据集的总共276个GEP(图5A)。
GEODB:该数据集包含13种不同化合物的GEP,获自从基因表达综合数据(GEO)获得的9个独立表达组(表1)。每个表达组具有至少6个DMSO对照和6个用于化合物处理的样品。3个表达组是针对MCF7乳腺癌细胞系(GSE9936-3化合物、GSE5149和GSE28662-2化合物)分析的,两组是针对MDA-MB-231转移性乳腺癌系(GSE33552-2化合物)分析的。其余的表达组是在B细胞淋巴瘤细胞系中分析,所述B细胞淋巴瘤细胞系是慢性淋巴细胞白血病患者来源的细胞系(GSE14973)、K422非霍奇金淋巴瘤细胞系(GSE7292)、裂解容许类淋巴母细胞系(GSE31447)、弥漫性大B细胞淋巴瘤患者衍生的细胞系(GSE40003)和套细胞淋巴瘤细胞系(GSE34602)。
DP92:该数据集含有在化合物处理后6、12和24hrs于3种不同B细胞淋巴瘤细胞系(OCI-LY3,OCI-LY7和U-2932)中分析的92种不同的FDA批准的晚期实验的工具性化合物的GEP。使用IC20以24hrs浓度处理化合物。在3个时间点中的每一个时间点使用DMSO作为对照介质,得到总共857个GEP。
为了运行DeMAND,产生了包括蛋白质-DNA相互作用和蛋白质-蛋白质相互作用的背景特异性基因调控网络(参见表2)。这些网络的背景特定信息是从源自相同背景的GEP获得的,而与背景无关的信息从大量的实验和计算证据获得。最后,朴素贝叶斯分类器被用来整合各种证据的相互作用,以获得最终的相互作用组(interactome)。下面提供关于如何产生相互作用组的详细描述。为了产生U133p2人B细胞相互作用组,使用了226个GEP,而对于U95av2人B细胞相互作用组,使用了254个GEP。为了产生乳腺癌相互作用组(BCI),使用从CMAP2数据集获得GEP。该数据集含有3,115个MCF7细胞系的特征谱。该数据集里的这些特征谱中有许多显示对处理没有反应,因此导致高冗余。为了减少这种冗余,通过随机比较2个对照(DMSO)样品产生具有大于2倍变化的基因的数目的背景变异分布。然后对于每次处理,如果与来自相同批次的相应对照样品相比显示出至少2倍的变化差异的基因数量小于根据背景分布确定的阈值,则滤出样品。这导致选择最终用于生成BCI的448个样本。
表2
表2:网络;*PDI:转录因子(蛋白质)-靶(NDA)相互作用;**PPI:蛋白质-蛋白质相互作用
对于通过网络中的相互作用连接的基因对,将其在给定条件(处理或对照)中的表达转化为二维概率密度。为了允许非线性,通过将化合物扰动和对照样品一起采用,对基因的表达数据进行等级转换。使用高斯核平滑,通过使用Silverman方法用以该点为中心的二维高斯概率密度替换点来估算概率密度。来自对应于化合物扰动的点的高斯概率密度的总和提供了扰动概率密度P,而来自对应于对照样品的点的高斯分布的总和提供了对照概率分布Q。在由样品数量定义的秩空间中的每个整数网格点处评估分布,并将其针对1的总和进行标准化,以创建有效的离散概率分布。
使用如下定义的Kullback-Leibler散度(KLD)来评估两个离散概率分布之间的距离:
KLD通过计算KLD(P|Q)和KLD(Q|P)来进行系统化,并对它们取平均值
KLD值的统计显著性使用由从随机基因对(无论它们是否共享网络边缘)估算的105个KLD值产生的零分布来确定,提供网络边缘的失调的p值。
通过使用网络中的相互作用的失调评分,评价了由基因施加的调控变化。这可通过使用费希尔方法组合从该基因周围的相互作用的KL-发散获得的p值来完成,所述费希尔方法将从给定的基因周围的k个相互作用获得的一组k个p值,pvi(i=1..k),转换成卡方统计
然后使用具有2k个自由度的卡方分布来计算组合的p值。使用费希尔方法组合p值的基本假设之一是独立性,即,假定围绕基因的相互作用是独立的,因此,在化合物扰动之后是失调的这些相互作用的p值也是独立的。由于给定基因a周围的相互作用共同具有该基因,因此这些相互作用之间的依赖性不能排除。因此,Brown方法被应用于p值依赖性的校正,其利用来自原始数据的协方差矩阵来校正从卡方统计获得p值所需的方差和自由度。因为人们必须校正相互作用之间的依赖性,而不是基因之间的依赖性,因此使用来自对共同基因a的线性拟合的残差之间的协方差(参见图12A)。这种校正消除了与网络中大量相互作用相关的基因失调的p值估算中的偏差(参见图12B)。具体来说,卡方的方差可以重新定义为
其中ρij是基因i与基因j的残差之间的相关性,以及
通过使用该方差,可将自由度重新定义为
并且人们可使用其来获得校正的p值,所述p值被使用Benjamini-Hochberg程序针对多个假设测试进行了校正。
化合物的直接靶标的知识从DrugBank数据库、MATADOR数据库和文献获得。从MATADOR数据库,注释为“直接”或“直接-间接”的基因被认为是化合物的已知靶标,而标记为“间接”的基因被丢弃。有关本研究中使用的化合物的靶标的列表。
进行子采样分析以用不同数量的GEP评估DeMAND的性能。这通过首先从DP14数据集里的每种化合物的18个化合物处理的样品和来自24个对照样品的相同数目的i个样品随机取样i(i=3...18)个样品,并对这i个样品进行DeMAND来完成。对于每种化合物和每个i,将其重复10次,并且将从子采样数据获得的10个结果与使用McNemar检验利用样品获得的结果进行比较。McNemar检验检查两个结果之间的同质性,同时考虑到所考虑的两个结果获自重叠样本的事实。
为了测试通过DeMAND预测为相似的化合物对是否也享有类似的治疗类别,使用来自解剖治疗化学(ATC)分类系统的每种化合物的药理学/治疗信息。具体来说,使用描述每种化合物的药理学/治疗亚组信息的第二级ATC分类。如果给定的化合物对共享相同的代码,则认为它们共享相似的治疗类别。
在DP92数据集里的92种化合物中,其中10种也在CTD2中进行了分析。在这10种化合物之间的所有潜在的化合物组合对中,5个化合物对没有相同细胞系的灵敏度曲线,因此不能获得它们之间的相似性。对于剩余的40个化合物对,使用至少23个共同细胞系至最多237个细胞系从灵敏度曲线获取相似性。通过灵敏度谱的Pearson相关性测量化合物之间的相似性,并且其显著性通过t-分布来估算,自由度等于共同细胞系的数目-2。
为了评价两种化合物的DeMAND预测之间的相似性,选择每种化合物的DeMAND结果中显著的DPG(FDR≤0.1)。然后使用这些选择的基因应用费希尔精确检验以计算它们之间的重叠的显著性。发现大量的常见基因在化合物之间没有显著的p值,这导致在费希尔精确检验中化合物对之间阴性基因组的高重叠,从而导致相似性估算中的偏差。为了校正该偏差,从背景组中扣除在整个化合物小组中从未显示为MoA的一部分的显著评分的基因,并重新计算费希尔精确检验。该校正对从最相似至最不相似的化合物对的分级没有影响,而是仅通过提供经校正的p值对相似性估算有影响。
为了获得DP92数据集中的每个化合物对的相似性的p值,独立地计算3个细胞系的相似性的p值,并且使用费希尔方法来组合这些p值。
为了评估内在网络灵敏度的变化是否对DeMAND的性能有影响,进行了梯度分析。为此,从网络中随机地逐渐去除相互作用,并且使用费希尔精确检验将DeMAND的输出与使用网络获得的输出进行比较。独立地对来自DP14数据集的14种化合物进行这种检验。由于计算约束,以逐步方式去除相互作用,首先通过去除10%的相互作用,随后去除另外10%的相互作用,并继续直至在网络中仅剩下10%的相互作用。
对于每种化合物和每种方法(DeMAND或t检验),将真阳性和假阳性率计算为排在前面的基因的分数的函数。对于具有d个直接靶标的化合物,通过方法考虑具有最显著的p值的前n个基因,真阳性率(TPR,也称为灵敏度)被定义为通过所述方法预测的该化合物的已知直接靶标的分数,并且假阳性率(FPR)被定义为在相同n个基因中为未知的直接靶标的基因的分数。
其中p是通过所述方法在前n个预测中预测的化合物的直接靶标的数目,N是在给定GEP数据集里分析的基因数目的总数。由于DeMAND为网络中的基因提供p值,因此当达到网络大小时,终止两个率的计算,并且假设曲线继续使用随机秩分配至其中两个率均等于1的终点。这在ROC曲线中描绘为连接达到网络大小时的点的直线,其中理论最大TPR和FPR值为1。
注意,在GEODB中,不同的表达集从不同的平台获得,因此可以预测的基因的总数和网络中的基因的数量二者均可变化。为了获得平均灵敏度曲线(在两个数据集里),我们因此根据该平台中总的可用基因中的排在前面的基因的分数(而不是使用排在前面的基因的数量)对真阳性和假阳性率求平均值。
弥漫性大B细胞淋巴瘤(DLBCL)细胞系OCI-LY3和OCI-LY7获自University Health Network(Toronto,Canada);U-2932DLBCL细胞系购自Leibniz-Institute DSMZ German Collection of Microorganisms and Cell Cultures;U-2-OS骨肉瘤细胞系获自ATCC(Cat#ATCC HTB-96)。将OCI-LY3、OCI-LY7、U-2932细胞系在37℃于补充有10%胎牛血清的Iscove改良的Dulbecco培养基(IMDM)中在5%CO2气氛中培养,而将U-2-OS细胞在补充有10%胎牛血清的McCoy's 5A培养基中进行培养。
基于它们在FDA批准的后期实验的和工具性化合物的初步筛选中的活性选择化合物。将OCI-LY3、OCI-LY7和U-2932细胞以100μL总体积以5×104个细胞/孔的密度接种在白色的组织培养物处理的96孔板中,使用Janus自动化液体处理系统(Perkin Elmer Inc.)。在37℃下培养12小时后,使板冷却至室温,然后通过Janus加入化合物。将化合物作为1μL的DMSO储液转移至测定板(基于板的一式三次重复)中,随后将其置于定轨振荡器上5分钟,然后放回培养箱中。24小时后,从培养箱中取出板并平衡至室温,然后每孔添加50μL CellTiter-Glo发光细胞活力测定(Promega Corp.)。将板在定轨振荡器上振荡5分钟,之后在Envision(PerkinElmer Inc.)中数据采集(0.5秒读取时间,增强发光)。使用IDBS Activity Base,利用四参数拟合模型测定IC20值。
对于每种化合物,通过在DMSO中稀释产生24hrs时的IC20的储液浓度。使用Janus自动化液体处理系统(Perkin Elmer,Inc.)将细胞以5×104个细胞/孔的密度接种在组织培养物处理的96孔板中,并用每种化合物的24小时的IC20在37℃,5%CO2下于加湿条件下处理6、12和24小时。对于每种化合物/条件组合,分析一个单一数据点,并将0.2%DMSO媒介物处理的样品用作对照。平行进行活力测定以监测化合物的效力。
在Janus自动液体处理系统(Perkin Elmer Inc.)上用RNAqueous-96Automated试剂盒(Ambion)分离总RNA,通过NanoDrop 6000分光光度计定量,并通过Agilent Bioanalyzer检查质量。使用基于标准T7的扩增操作方案,使用Illumina TotalPrep-96RNA扩增试剂盒(Ambion)将300ng RIN值>7的样品的每一种转化为生物素化的cRNA,并在人类基因组U219 96-阵列板(Affymetrix)上杂交。根据制造商的操作方案在GeneTitan仪(Affymetrix)上进行阵列板的杂交、洗涤、染色和扫描。
靶向每个指定基因的小干扰RNA(siRNA)和siControl SMARTpools获自Dharmacon Technologies(参见表3)。通过制备1mL Opti-MEM(Invitrogen),6μL Lipofectamine-RNAiMAX试剂(Invitrogen)和1.25μL的10μM RNAi溶液(最终工作浓度为6.4nM)的溶液,并将混合物(1mL/孔)在6孔皿中于37℃下温育20分钟来进行反向转染。将U-2-OS细胞从培养瓶中分离,并以4×105个细胞/mL的密度重悬浮于2X含血清的培养基中。将1mL细胞悬浮液转移至含有转染混合物的每个孔中,并将6孔板放回培养箱中。48小时后,分离细胞,并使用相同的程序再次反向转染。48小时后,将细胞进行胰蛋白酶化并重新接种至384孔板中以测定对长春花新碱的灵敏度。在长春花新碱处理48小时后,将10μL的在U-2-OS生长培养基中的50%阿尔玛蓝(Life Technologies)溶液转移至384孔板中,得到10%终浓度的阿尔玛蓝。将板再温育16小时以允许还原阿尔玛蓝,这导致红色荧光的产生。使用Victor 3板阅读器(Perkin Elmer,Inc.)测定荧光强度,并将其用于计算生长抑制百分比。
表3
表3;用于长春花新碱MoA的siRNA
将细胞在盖玻片上生长至~50%汇合,并用化合物处理24小时。用3.7%甲醛的PBS溶液将细胞固定15-30分钟,然后用PBS洗涤5次。细胞膜用0.2%Triton-X的PBS溶液透化10分钟,并用TBS(10mM Tris[pH7.5],150mM NaCl)漂洗一次。将透化的样品用10%山羊血清的TTBS(0.1%Tween-20的TBS溶液)溶液封闭30-60分钟,并用TTBS洗涤一次。在室温下用在1%山羊血清的TTBS溶液中的抗微管蛋白抗体(Santa Cruz cat#sc-32293)探测微管网络30-60分钟,然后在TTBS中洗涤10分钟。使用Alexa Fluor抗小鼠抗体(Invitrogen,目录号A-11005)作为二抗,以使用共聚焦显微镜的60x透镜使微管网络可视化。
如下进行GPX4酶活性测定。简言之,将1×106个细胞重悬于细胞裂解缓冲液中。使用超声处理制备细胞裂解物,然后以14,000rpm离心10分钟。使用Bradford蛋白质测定(Bio-Rad)来测定澄清的细胞裂解物的蛋白质浓度。将200微克细胞蛋白质与磷脂酰胆碱氢过氧化物(PC-OOH)(GPX4特异性底物)和还原型谷胱甘肽(GPX4辅因子)混合。将混合物在37℃下温育30分钟,然后使用氯仿:甲醇(2:1)溶液进行脂质提取。使用旋转蒸发器蒸发脂质提取物,并重新溶解于100%乙醇中,然后注射入LC-MS仪以用于PC-OOH定量。
将U-2932细胞接种在10cm培养皿(2×106/皿)中,并在37℃下生长16小时。用媒介物(0.4%DMSO),1mM柳氮磺吡啶(24小时IC50)或1mM六甲蜜胺(24小时IC50的双倍)处理细胞,并温育24小时。然后收获细胞,沉淀,用含有1mM EDTA的400μl冰冷的PBS洗涤一次并超声处理。在沉淀并除去碎片后,使用QuantiChrome谷胱甘肽测定试剂盒(BioAssay Sytems)在技术上一式三份定量120μL样品中的氧化和还原型谷胱甘肽。将谷胱甘肽量相针对用Bradford测定法(Bio-Rad)测量的蛋白质浓度标准化。
将U2932细胞(2x105)接种在6孔板中,并在37℃下培养16小时。将细胞用测试化合物处理指定的时间,然后收获,沉淀并用PBS洗涤一次。对于脂质ROS检测,将细胞用含有C11-BODIPY(581/591)(2μM)(Life Technologies)的Hanks平衡盐溶液(HBSS,Life Technologies)重悬,并在37℃温育10分钟。然后将细胞沉淀,重悬于500μL HBSS中,通过40μM细胞过滤器(BD Falcon)过滤,并使用BD Accuri C6流式细胞仪(BD Biosciences)分析。使用FL1通道测量C11-BODIPY信号。以生物学一式三份重复进行实验,并且显示了代表性结果。
JAK2选择性抑制剂TG101348和丝裂霉素C分别购自Selleckchem和Tocris Bioscience,将其溶解于DMSO中。在96孔板中用所示化合物处理OCI-LY3细胞,并使用CellTiter-Glo发光细胞活力测定(Promega Corp)测定生长。通常,在所期望的化合物存在或不存在(仅DMSO)的情况下,将200μL生长培养基中每孔30,000个OCI-Ly3细胞生长48小时,然后根据制造商的说明书使用CellTiter Glo进行测定。
出于说明而非限制的目的,参考图3,DeMAND可接受与来自对照样品和扰动样品的基因表达谱相组合作为调控网络的输入。通过使用该输入,网络中的每个相互作用被单独地测试失调。这通过首先使用高斯核方法对参与相互作用的两个基因的共表达散布图进行平滑(即用围绕该值的二维高斯代替二维图中的每个表达值),从而获得内在概率密度的估算来进行。使用KL散度评价对照和扰动样品的概率分布之间的差异,并且通过将该值与随机基因对的KL散度值进行比较来测定该差异的统计显著性。接下来,通过整合其相互作用的p值,同时考虑它们之间的依赖性来测定网络中每个基因的失调(也参见图12),从而提供反映基因失调的显著性的单一评分。作为输出,DeMAND提供输入数据中的基因和与其失调相关的p值的列表。
为了说明而不是限制的目的,图4A显示了通过DeMAND(左)和通过t检验(右)预测的DP14数据集里的每种化合物的已知的直接靶的最佳等级。DeMAND在11种化合物中的10种中鉴定了处于更好等级的已知靶。
为了说明而不是限制的目的,参考图4B,如使用来自每个时间点一起的(例如,所有数据)GEP或使用单独的每个时间点(6小时、12小时或24小时)的GEP通过DeMAND预测的DP14数据集里的每种化合物的已知的直接靶的标准化的最佳等级。通过取四个预测(所有数据,6小时、12小时或24小时)中的最小秩的比率和四个预测中的每一个的秩,对每种单独的化合物进行标准化。每个圆的尺寸/颜色表示标准化的秩,其中较大或较暗的圆代表较低等级或较好的预测。该图显示了仅针对具有FDR≤0.1的预测的标准化的秩。
为了说明而不是限制的目的,图4C显示了作为假阳性率的函数的平均真阳性率(灵敏度),其用于使用DeMAND预测(401)和使用t检验分析(402)来鉴定DP14数据集中的化合物的已知的直接靶。对于所示的每个假阳性率值,DeMAND获得比t检验更高的真阳性率。DeMAND预测的右上方的直线表示不在调控网络中的基因的随机秩分配。
为了说明而非限制的目的,参考图4D,blebbistatin是参与整联蛋白信号传导的肌球蛋白II的特异性抑制剂,其是非肌肉肌球蛋白。在B细胞中,肌球蛋白II对于B细胞受体信号传导和免疫突触稳定性、细胞与细胞接触、趋化性、细胞周期和细胞分裂至关重要。Pyk2/PTK2B是调节肌球蛋白II磷酸化的关键激酶,并且被整联蛋白或趋化因子介导的Ca+2信号传导以及应激中的活性氧(ROS)生成所激活,导致通过GRB2衔接蛋白基因的Src家族激酶缔合(FYN)。虽然DEMAND不将肌球蛋白II预测为blebbistatin的MoA,但其将PTK2B、FYN和GRB2预测为MoA基因。
为了说明而非限制的目的,图4E显示了GSEA图,其显示了已知影响MTOR的化合物(即雷帕霉素(左上和左下)和放线菌(右上和右下))的雷帕霉素响应基因集合的富集。当根据DeMAND预测(上)对基因进行分级时,两种化合物的富集是统计上显著的。当使用t检验预测进行分类时,对于雷帕霉素(左下)没有观察到显著性,而对于放线酮(右下)统计上显著性仍然存在。
为了说明而不是限制的目的,图5A显示了使用DeMAND(501和502)和使用t检验分析(503和502)鉴定DP14数据集中的所有化合物的已知直接靶的平均灵敏度(真阳性率),其为排在前面的预测的数量的函数。对于许多排在前面的基因,DeMAND优于t检验分析。例如,DeMAND在前100个预测中实现了近15%的灵敏度,而t检验只实现了3%的灵敏度。此外,通过t检验分析鉴定的大多数直接靶标也被DeMAND鉴定,如通过缺乏红色(直至选择超过400个排在前面的基因)所证明的。相反的情况是不真实的,DeMAND鉴定许多未被t检验鉴定的直接靶标,如由大的蓝色区域所示。
为了说明而不是限制的目的,图5B显示了比较三种拓扑异构酶抑制剂-喜树碱、多柔比星和依托泊苷的MoA的示意图,其聚焦于通过DeMAND预测参与每种化合物的MoA的基因。对于多柔比星是独特的基因在具有橙色背景的区域(509)中,而对于喜树碱和依托泊苷是独特的基因在具有紫色背景的区域(510)中。所述化合物共享主要的DNA损伤修复MoA,包括GADD45A、PCNA和CDNK1A。多柔比星MoA包括参与组蛋白回收的KAT5和参与维持心脏祖细胞的SIK1,并因此将多柔比星与其最显著的心肌病的治疗限制性副作用联系起来。
为了说明而不是限制的目的,图5C显示了DeMAND预测的参与DNA损伤应答的基因的分级。DeMAND预测GADD45A(504)(规范的DNA损伤诱导基因)及其众所周知的相互作用基因CDKN1A(503)、PCNA(505)、CCNB1(508)、AURKA(507)和PLK1(506),具体而言,DNA损伤剂中的5种(即喜树碱、多柔比星、依托泊苷、丝裂霉素C和长春花新碱)的统计上显著的MoA推断基因。
为了说明而不是限制的目的,图6A显示了用DMSO、长春花新碱、非特异性siRNA和靶向RPS3A的siRNA处理的细胞的通过免疫组织化学进行的微管网络的可视化。与DMSO对照相比,非特异性siRNA对微管网络没有影响。长春花新碱和siRPS3A都显著改变微管网络。U-2-OS细胞用4nM长春花新碱处理24小时。
为了说明而非限制的目的,图6B显示了对于用非特异性siRNA(602)、靶向CCNB1(604)、VHL(601)、NFKBIA(603)和RPS3A(605)的siRNA处理的U-2-OS细胞,作为长春花新碱浓度的函数的相较于未经长春花新碱处理的siRNA转染的细胞的细胞抑制百分比。RPS3A和CCNB1的抑制降低细胞对长春花新碱治疗的灵敏度,而VHL的抑制使灵敏度增加至两倍。
为了说明而不是限制的目的,图6C显示了对于单独用丝裂霉素C(空心圆圈)或用与JAK2抑制剂(使用0.2uM(实心圆圈)、0.4uM(倒三角形)和0.6uM(正方形))组合的丝裂霉素C处理的OCI-LY3细胞,作为丝裂霉素C浓度的函数的相较于DMSO处理的细胞的细胞抑制百分比。JAK2抑制剂水平与细胞对丝裂霉素C处理的灵敏度降低相关。
为了说明而不是限制的目的,图7A显示了当使用U95av2调控网络时,DP14数据集中的每种化合物的显著的DeMAND结果的GSEA曲线,其中分级根据使用U133p2调控网络的结果。化合物显示出统计显著性(p值≤10-9),例证了DeMAND结果对输入网络不敏感。
为了说明而不是限制的目的,参考图7B,在从网络中随机除去相互作用之后,将DeMAND应用于DP14数据集,并且使用费希尔精确检验将显著结果与原始结果进行比较。该图显示了作为除去的边缘的百分比的函数的费希尔精确检验的log 10的p值。在除去超过60%的相互作用之后,重叠的p值低于统计显著性(在多个假设校正之后)。
为了说明而不是限制的目的,参考图7C,使用对照和扰动数据的自举子集将DeMAND应用于DP14数据集,并且使用18个样品和18个对照样品的结果的差异的显著性使用McNemar检验(其中大的差异提供显著的p值)来评估。该图显示作为样本大小的函数的log 10的p值。对于6个和以上的样本大小,p值在使用18个样品的结果的单一标准偏差内。
为了说明而不是限制的目的,图8A显示了通过DeMAND(左)和通过t检验(右)预测的GEODB数据集里的每种化合物的已知直接靶的最佳等级。
为了说明而非限制的目的,图8B显示了作为假阳性率的函数的平均真阳性率(灵敏度),其用于使用DeMAND预测(801)和使用t检验分析(802)鉴定GEODB数据集里的化合物的已知直接靶。对于假阳性率的大多数值,DeMAND实现比t检验更高的真阳性率。DeMAND预测的右上方的直线表示不在调控网络中的基因的随机秩分配。
为了说明而非限制的目的,图8C显示了使用DeMAND(803)和使用t检验分析(804)的GEODB数据集中的所有化合物的已知直接靶的平均真阳性率(灵敏度),其为排在前面的预测的数量的函数。对于许多排在前面的基因,DeMAND优于t检验分析,在前100个预测中达到几乎15%的灵敏度,而t检验只达到2%的灵敏度。
为了说明而不是限制的目的,参考图9A,以检测化合物之间的相似性,DeMAND首先推断每种化合物的MoA。使用费希尔精确检验的列表之间的重叠的显著性来评价化合物对之间的相似性。
为了说明而不是限制的目的,参考图9B,通过共享已知直接靶标的每对化合物的精密度(903)来评估DP92数据集中的92种化合物之间的预测的相似性的排名,根据CTD2数据(904)产生类似的灵敏度分布,根据解剖治疗化学(ATC)分类(902)或上述证据(901)共享相同的分类。结果表明,预测的相似性在区分具有相似MoA的化合物中是成功的。
为了说明而不是限制的目的,参考图10A,DeMAND预测的化合物对相似性用具有支持性外部证据的化合物对来进行富集。根据其显著的DeMAND预测的重叠的显著性(通过费希尔精确检验)对化合物对进行分类,并且具有外部证据的一组配对由共享相同ATC类别或共享确定的直接靶标的化合物对组成,或者使用CTD2数据显示显著相关性。
为了说明而不是限制的目的,图10B显示了作为灵敏度的函数的精度(真阳性率),其中阳性由共享相同直接靶标,或者共享相同的ATC分类,或者共享根据CTD2数据集跨多个细胞系的相似的灵敏度曲线的对确定。所述曲线是针对通过显著的DeMAND预测的重叠(1001)、通过t检验分析的重要基因的重叠(1003)预测的化合物相似性和通过MANTRA算法(1002)计算的相似性的。T检验分析实现较差的精密度,并且DeMAND对于大多数灵敏度值达到最佳精密度。注意,t检验线结束短(灵敏度为0.14),因为许多化合物对显著差异表达的基因的没有重叠。
为了说明而不是限制的目的,图10C显示了CTD2数据集中的化合物相似性与通过显著的DeMAND预测的重叠估算的化合物相似性之间的比较。DeMAND相似性预测提供0.59(p值=7.8×10-5)的与CTD2结果的显著Spearman相关性。
为了说明而不是限制的目的,图10D显示了CTD2数据集中的化合物相似性与通过MANTRA算法估算的化合物相似性之间的比较。MANTRA相似性预测提供0.26(p值=0.11)的与CTD2结果的非显著Spearman相关性。
为了说明而非限制的目的,图10E显示了CTD2数据集中的化合物相似性与通过由t检验显著差异表达的重叠估算的化合物相似性之间的比较。T检验相似性预测提供0.39(p值=0.013)的与CTD2结果的边际显著的Spearman相关性。应当注意,多种化合物没有重叠,因此全部等级相同。
为了说明而不是限制的目的,图11A显示在阴性对照(DMSO,1101)、阳性对照、柳氮磺吡啶(1102)和六甲蜜胺(1103)处理细胞后的GSH浓度显示,柳氮磺吡啶处理降低活性GSH的水平(相较于对照),而六甲蜜胺处理导致与对照无法区分的活性GSH的水平。
为了说明而不是限制的目的,图11B显示如通过无细胞裂解物(1104)、利用未处理的细胞裂解物(1105)和来自用六甲蜜胺(1106)处理的细胞的细胞裂解物的质谱法测量的GPX4-特异性底物PC-OOH的水平。当细胞用六甲蜜胺处理时,PC-OOH的水平类似于无裂解物,并且与未处理的裂解物显著不同,表明六甲蜜胺处理降低了GPX4的活性。
为了说明而非限制的目的,图11C显示通过对照DMSO处理的细胞(1107)或化合物处理后的细胞(1108)的流式细胞术测量的脂质反应性氧化物质(ROS)的水平。两种化合物的处理显著升高脂质-ROS的水平,例示了六甲蜜胺处理的功能效应类似于柳氮磺吡啶的功能效应。
为了说明而非限制的目的,参考图11D,已知柳氮磺胺吡啶通过系统xc-胱氨酸/谷氨酸反向转运体起作用,对谷胱甘肽(GSH)和GPX4产生下游作用,并导致脂质反应性氧化物质的积累。DeMAND预测柳氮磺吡啶与六甲蜜胺之间的显著相似性,但并不预测六甲蜜胺对GSH的失调,而是预测GPX4的失调。该预测通过图(A)-(C)中的实验结果得到验证。
为了说明而非限制的目的,参考图12A,组合p值的Brown方法需要估算导致p值的结果之间的相关性。为了估算涉及相同基因(A)的两个相互作用之间的相关性,计算来自与A(基因B和C)相互作用的每个基因的线性拟合的残差,并且接下来计算残差之间的相关性。
为了说明而不是限制的目的,参考图12B,对于DP14数据集里的化合物,将基因失调的p值的log10的概率密度作为所述基因所参与的相互作用数(程度)的函数作图。校正前(左框)的概率分布取决于基因的程度,但在校正后(右框),这种偏差被去除。
实验程序。
DeMAND可在网络ε和基因表达集GEP中用作输入,将其分为对照集GEPc和扰动集GEPp。每组含有N个基因以及分别地Mc和Mp个样品的测量,给出总共M个样品。
该算法通过网络中的每个相互作用(其中1≤i,j≤N)运行,并估算分别获得基因Gi和Gj的表达值Ei和Ej的联合概率分布。为此,首先对表达值进行秩变换,接着进行高斯核平滑,其中,对于样品1≤m≤M的每个表达点(Eim,Ejm)用围绕该点的2维高斯取代。高斯的标准偏差σi和σj通过Silverman经验法则获得,给出其中是数据的标准偏差。然后通过对它们相应样品的高斯进行求和,创建两个概率分布,一个用于对照样品,一个用于扰动样品。然后在每个整数点(k,l)对分布进行采样,其中1≤(k,l)≤M,给出
将这些离散概率分布标准化以确保超过k和l的和等于1。
给定两个概率分布,一个用于对照样品,一个用于扰动样品,计算它们之间的Kullback-Leibler散度(KLD)以评价两个分布之间的差异。当尝试使用一个概率分布来近似另一个概率分布时,这可以被解释为信息的丢失。在这种情况下的KLD采取形式:
为了确保距离的对称性,计算以下等式
这可以提供网络中每个边缘的失调的测量。
零假设是边缘包括其相互作用不受扰动影响的两个基因。因此,为了创建保持这种假设(也称为零分布)的期望值的分布,随机选择两个基因并计算它们的KLD。注意,这两个基因通常不共享网络边缘,因此它们的表达水平之间没有关系。因此,观察到的使用对照与扰动样品之间的这种关联性的变化(通过其概率分布的变化来测量的),因此应该是偶然的。
重复该过程105次,从而创建零模型,然后将其用于估算获得给定KLD值(或更高)的概率。这通过计算在零分布中获得这样的值或更高值的次数并将其除以所述零分布中的值的总数目来进行。这被称为KLD的p值,表示为Pvij。
为了评估每个基因Gi的失调,对其中其所涉及的所有相互作用的p值进行积分。这是使用费希尔方法对p值积分来完成的,以使得p值被对数变换和求和以得到卡方统计
经过积分的p值从具有2k自由度的卡方分布获得,其中k是被积分的相互作用的数量。
对多个p值进行积分的费希尔方法需要从独立实验获得这些p值。然而,在网络的情况下,这不是正确的,并且与共同基因的多种相互作用可以是相关的。为了校正该偏差,使用了改良的Brown方法。具体来说,卡方的方差被定义为
其中ρij是基因i和基因j的残差之间的相关性,以及
使用该方差,将自由度重新定义为
注意,此处,需要的是相互作用之间而非基因之间的相关性的估算。为了估算相互作用与之间的相关性,估算来自作为基因Gi的函数的基因Gj和Gk的独立线性拟合的残差,并使用这些残差之间的相关性作为相互作用之间的相关性的替代。这是假设如果两个相互作用是独立的,则残差不应该相关,并且残差之间的相关性是由于这两个相互作用具有共同基因的事实。所得p值用作基因Gi失调的显著性的估算。
为了建立组织特异性相互作用组,可使用贝叶斯证据积分方法(BEIA)。朴素贝叶斯分类建立了特定相互作用的概率可使用该类相互作用的先验概率和支持其的每条线索的似然比(LR)的乘积来计算。该方法需要蛋白质-蛋白质相互作用(PPI)和蛋白质-DNA相互作用(PDI)的阳性和阴性实例的大的数据集(称为金标准阳性和阴性集(分别为GSP和GSN))来训练每条线索。根据朴素贝叶斯分类器的要求,这些证据源中的每一个可以事先被评估为独立于其余的。
为了预测PPI,可以整合以下证据来源。
四个真核生物的数据库IntAct、BIND和MIPS的分子相互作用
人高通量筛选
GeneWays文献数据挖掘算法
基因本体论7(GO)生物过程注释和Interpro蛋白质结构域注释
同样,来自相关组织类型的GEP集合的共表达数据可在每个相互作用组中引入背景特异性。
PPI的GSP是来自HPRD、BIND和IntAct数据库的概要,其包含一组包含9,839个基因的48,648个独特PPI(在去除同源二聚体后)。阴性相互作用GSN的组定义为来自存在于不同细胞区室中的GSP编码蛋白质中的基因的基因对,其代表一组5,362,594个阴性基因对。先前估算在基因组中的≈22,000个基因之间存在≈300,000个PPI相互作用,这意味着在800个潜在相互作用中有1个发生。因此,具有似然比大于800的相互作用表明其为真阳性的概率为50%。
为了预测PDI,可以收集以下证据来源:
来自TRANSFAC的小鼠相互作用
BIND数据库
在靶基因的启动子中鉴定的TF结合位点(TFBS)
GeneWays文献数据挖掘算法
通过ARACNe算法从每种组织类型中的GEP推断的PDI可以引入背景特定性信息。
为了产生PDI的GSP,从Professional(TRANSFAC)、BIND和Myc(MycDB)数据库提取人的相互作用。这导致包括585个TF和2034个靶标的4500个相互作用的GSP PDI集。随机产生GSN,其含有由TF和靶标组成的100,000个基因对,排除了其中两个基因参与GSP相互作用或由基因本体(Gene Ontology)定义的相同生物过程的对。
基于ARACNe网络中TF的靶标数目,为每个TF定义PDI的阈值。例如,在≈22,000个潜在靶标中由ARACNe预测的具有100个靶标的TF的先验将是100/22000,这将指示LR>220。然而,如果产生的截断值小于5(意味着该TF调节超过基因中的第5个),则无论如何使用LR>5。
此外,报告的相互作用组还引入来自涉及在每种细胞背景中表达的基因的两个GSP的相互作用对。
前述内容仅仅举例说明所公开的主题的原理。鉴于本文的教导,对所描述的实施方案的各种修改和改变对于本领域技术人员将是显而易见的。因此,应当理解,本领域技术人员将能够设计出许多技术,虽然这里没有明确地描述,但是这些技术体现了所公开主题的原理,因此在精神和范围内。
- 还没有人留言评论。精彩留言会获得点赞!