刀额新对虾中磷脂的亲水色谱‑串联质谱检测方法与流程
本发明属于食品检测领域,涉及一种刀额新对虾中磷脂的亲水色谱-串联质谱检测方法,具体指一种刀额新对虾中磷脂酰胆碱、磷脂酰乙醇胺、磷脂酰丝氨酸三大类磷脂的结构鉴定和定量分析方法。
背景技术:
刀额新对虾(Metapenaeus ensis),即基围虾,是我国养殖小型海产虾类之一。因其具有食性杂、生长快、耐高温、适盐范围广、抗不良环境能力强等优点,近几年在中南沿海地区广泛开展了人工养殖,显示出良好的经济前景。虾类水产资源脂质含量高,富含Ω-3族和Ω-6族多不饱和脂肪酸和磷脂酰胆碱(Phosphatidylcholine,PC)、磷脂酰乙醇胺(Phosphatidylethanolamine,PE)、磷脂酰丝氨酸(Phosphatidlserine,PS)等磷脂类物质。
磷脂是一类含有磷酸的脂类。磷脂结构主要由甘油骨架、极性基团和不同长度和饱和度的脂肪酸链组成。根据磷脂极性基团不同,可将磷脂分为磷脂酰胆碱、磷脂酰乙醇胺、磷脂酰丝氨酸等类别。生物体中存在的磷脂种数以千计,结构多样、种类复杂,具有独特的化学结构。磷脂是维持细胞结构的重要组成部分,也是新陈代谢不可或缺的营养元素。磷脂还具备广泛的生物学功能,诸如活化细胞,维持代谢与荷尔蒙分泌均衡,增强人体免疫力等。实验证明,磷脂可以分解血脂、胆固醇,阻止多余脂肪在血管壁沉积,使血管循环顺畅,被誉为“血管清道夫”。高效的刀额新对虾中磷脂分子的检测方法将有利于深化刀额新对虾中磷脂的营养学研究,促进刀额新对虾为原料的产品开发。
传统磷脂检测方法包括高效液相色谱(HPLC)、核磁共振波谱(NMR)、荧光光谱(FLR)等,这些方法分析耗时长、灵敏度低。亲水色谱(HILIC)是专门分离强极性和亲水性化合物的一种方法。因为使用常规反相色谱流动相,使亲水色谱具有较好的质谱兼容性。
目前现有的脂类分离常规方法为正相色谱,需要特殊的色谱设备与流动相(正己烷、异丙醇、氯仿),且该流动相所得的洗脱液无法与质谱设备直接联用。
技术实现要素:
本发明要解决的技术问题是提供一种操作简便、定性定量能力较强的刀额新对虾中磷脂酰胆碱、磷脂酰乙醇胺和磷脂酰丝氨酸的亲水色谱-串联质谱检测方法。
为了解决上述技术问题,本发明提供一种刀额新对虾中磷脂的亲水色谱-串联质谱检测方法,包括以下步骤:
1)、样品前处理:
将刀额新对虾肉研磨成虾糜,按照1g/15~20ml的料液比,在虾糜中加入有机溶剂Ⅰ后震荡混匀;然后进行超声(探头式超声)冰浴提取(提取时间为10~15分钟),提取完毕后加入纯水并震荡混匀,得混合物,每1g虾糜配用10~15mL的超纯水;有机溶剂Ⅰ为甲醇和氯仿的混合液,甲醇与氯仿的体积比为0.5~2:1;
混合物用冷冻离心机离心,从离心所得物中移取下相溶液;所述下相溶液低温(≤10℃)干燥(用氮气进行低温吹干)后,用有机溶剂Ⅱ复溶,得脂质粗提物;所述有机溶剂Ⅱ是体积浓度≥30%的甲醇水溶液、或甲醇(即,30~100%的甲醇水溶液,较佳为90%的甲醇水溶液),
2)、液相分离:
将脂质粗提物进行液相分离;色谱柱为YMC Triart二醇基HILIC柱(4.6×250mm,3μm)。色谱条件采用梯度洗脱法,梯度系统由流动相A和流动相B组成;其中流动相A为含有53mmol/L甲酸的乙腈溶液(pH约为4.0~4.5),流动相B为含有60mM甲酸铵和53mM甲酸的水溶液(pH约为3.6);
梯度系统为:0~15min,维持2%流动相A;15~60min,将流动相A从2%提高到40%;60~70min,维持40%流动相A;70~80min,将流动相A从40%降回初始状态的2%;上述%均为体积%;
备注说明:液相的洗脱液直接进入质谱分析;
液相色谱采用Agilent 1100系统,配以四元泵、自动进样器、色谱柱恒温箱等元件;
3)、将步骤2)所得的洗脱液进行质谱检测分析。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的改进:
所述步骤3):
采用QSTAR四级杆/飞行时间质谱仪,配以TurboIonSpray离子源;负离子模式检测;扫描范围为500~1000Da;去簇电压(DP)为50V;聚焦电势(FP)为200V;离子源电压为4500V;去簇电势2(DP2)为10V;气源1(GS1)为50psi;气源2(GS2)为45psi;气帘气(CUR)为40psi;温度为400℃;子离子扫描(PIS)采用的碰撞能为20V;数据采集与分析采用Analyst QS v2.0。
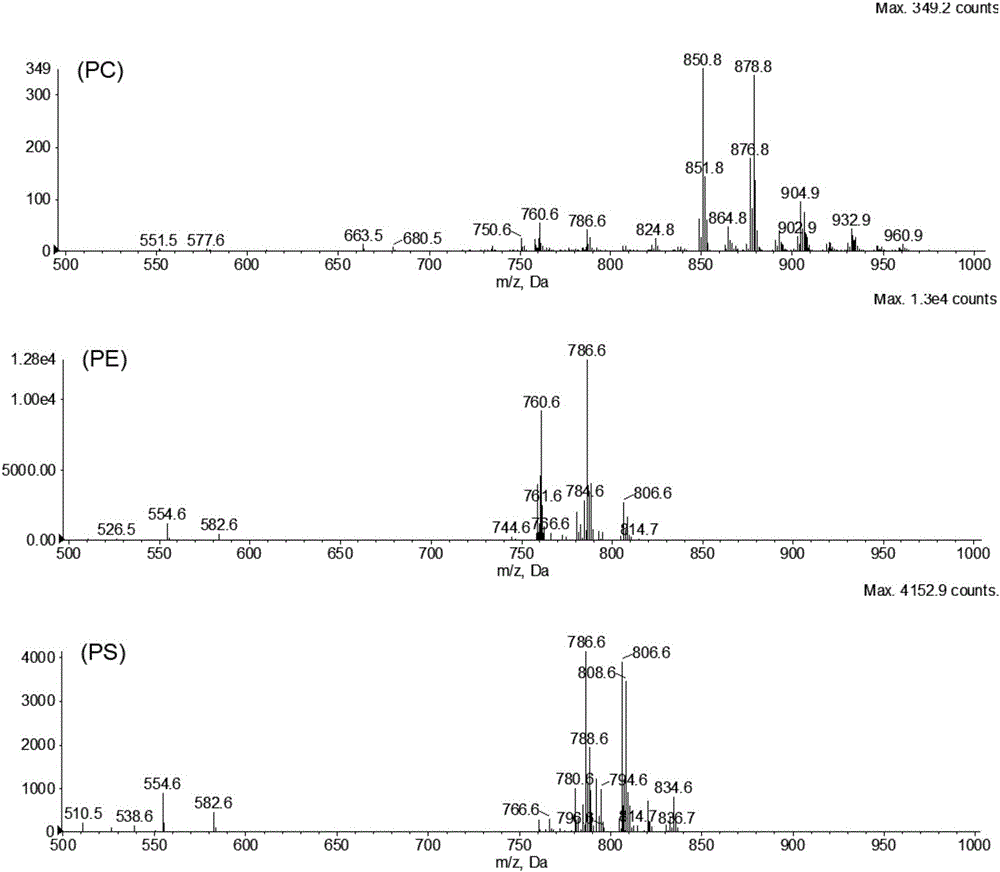
备注说明:液相色谱能将磷脂按类分离,如图1,每个峰代表一类磷脂(有些峰是杂质),每类磷脂中,按照碳链长度不同与双键数量不同,包含很多磷脂分子,质谱对每类磷脂检测后,如图2,可以得到每类磷脂的不同磷脂分子的质荷比(即质量与电荷之比,本实验中电荷为1,所以质荷比的值就是磷脂分子质量),每个峰代表一个磷脂分子种,根据磷脂质量,再经过lipidview搜库就可得到结构信息。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤1)中,以移取下相溶液后的离心所得物(即,包括上相溶液、位于上相溶液和下相溶液之间的固体物)替代虾糜,以有机溶剂Ⅰ中的氯仿替代有机溶剂Ⅰ,再重复进行提取1~3次;
合并所有提取步骤所得的下相溶液合并后进行后续的干燥和复溶。
备注说明:离心后离心管内溶液上下分层,刀额新对虾肉糜则呈饼状居于两相中间。
甲醇、氯仿、水三者在一起的时候不会混溶,会分成上下两相,甲醇极性较强会与水混溶,因此在离心管中,氯仿混溶少量甲醇在下相,水混溶多量甲醇在上相;由于移取走下相后,实际大部分甲醇依然在上相中并未被移取,因此重复提取的时候只需要加入氯仿即可。即,重复提取时氯仿的体积用量=机溶剂Ⅰ中的氯仿的体积用量。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤1)中超声条件为25kHz。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤1)中冷冻离心为于4℃以8000rpm离心15分钟。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤2)中,梯度洗脱时的流速为180~220μL/min(较佳为200μL/min)。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤2)中进样量(即,脂质粗提物的量)为5~10μL。
作为本发明的刀额新对虾中磷脂的亲水色谱-串联质谱检测方法的进一步改进:
所述步骤1)中,虾糜与复溶用的有机溶剂Ⅱ的料液比为1g/1~2ml。
本发明的步骤1)的样品前处理中:
取均质后的刀额新对虾肌肉组织样品(虾糜)于离心管,加入有机溶剂Ⅰ后震荡混匀。探头式超声冰浴提取,完毕后向离心管加入纯水并震荡混匀。混合物用冷冻离心机离心,离心后离心管内溶液上下分层,刀额新对虾肌肉组织样品则呈饼状居于两相中间。用移液枪将下相溶液转移到另一个新的移液管,继续向上相和样品中加入氯仿并重复上述操作提取1~3次(例如为两次)。将提取的有机相合并,用氮气低温吹干,并用甲醇水溶液复溶。
在本发明中,复溶后过0.22μm的PTFE滤膜,得脂质粗提物。
本发明采用二醇基HILIC柱,以53mmol/L甲酸的乙腈溶液为流动相A,60mM甲酸铵和53mM甲酸的水溶液为流动相B;经梯度洗脱后(0~15min,维持2%流动相A;15~60min,将流动相A从2%提高到40%;60~70min,维持40%流动相A;70~80min,将流动相A从40%降回初始状态的2%)将磷脂分离。相比常规色谱技术,该方法能使脂类分离在实验室常规仪器反相色谱中得到实现,有效避免了传统正相色谱所需的设备与流动相,使用甲醇或乙腈即可,并且可与质谱直接联用。
本发明的检测方法样品制备、液相分离、质谱检测,操作简便、简单易培训、定性定量能力较强、实用性强,适合实验室广泛使用。
综上所述,本发明建立的一种刀额新对虾中磷脂的亲水色谱-串联质谱检测方法操作简单快捷,灵敏度高,结果稳定可靠,对于水产品脂质组学发展具有重要意义。采用本发明的方法能准确检测出样品中PE、PS和PC的总含量,以及实现对PC、PE及PS三类磷脂中各个分子种进行质谱分析鉴定。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
图1为刀额新对虾肌肉组织脂质提取物的HILIC-TOF/MS检测图;
TIC of+TOF MS:from Sample of 20150610.wiff(Turbo Spray)。
图2位刀额新对虾脂质提取物中PC,PE和PS的质谱图;
上图(PC):
+TOF MS:20.176 to21.528min from Sample7of 20150610.wiff
a=3.59917355969297020e-004,t0=-5.28098098505142840e+000(Turbo Spray)
中图(PE):
+TOFMS:49.449to50.000min from Sample7of 20150610.wiff
a=3.59917355969297020e-004,t0=-5.28098098505142840e+000(Turbo Spray)
下图(PS):
+TOFMS:48.899to49.449min from Sample7of 20150610.wiff
a=3.59917355969297020e-004,t0=-5.28098098505142840e+000(Turbo Spray)。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
本发明中:
材料与试剂
三种磷脂标准品:PC-14:0/14:0、PE-15:0/15:0和PS-14:0/14:0纯度均大于99.9%(美国Avanti Polar Lipids公司);以甲醇为溶剂配置成1mg/mL的标准品母液。氯仿、甲醇、乙腈等试剂购买自德国Merck公司。电阻率为18.2MΩ·cm的超纯水过滤自Milli-Q系统(美国Millipore公司)。
实施例1、一种刀额新对虾中磷脂的亲水色谱-串联质谱检测方法
具体为依次进行以下步骤:
1)、样品前处理:
刀额新对虾去皮去排泄腺后,取腹部横纹肌肉组织作为虾肉并研磨成虾糜(浆状)。称量1.0g虾糜于5mL离心管,加入18mL氯仿/甲醇混合液(1:2,v/v)后震荡混匀。在250W下超声波(25kHz)冰浴提取10分钟后向离心管加入12.5mL超纯水并震荡混匀。混合物用冷冻离心机(美国Thermo Scientific公司)于4℃以8000g离心5分钟,离心后离心管内溶液上下分层,组织样品则呈饼状居于两相中间。用移液枪将下相溶液转移到另一个新的移液管,继续向上相和样品中加入6mL氯仿并重复上述操作提取两次。将三次提取的有机相(下相溶液)合并,用氮气低温吹干,并用甲醇水溶液2ml(甲醇:水=9:1,v/v)复溶后过0.22μm的PTFE滤膜。
2)、液相分离
液相色谱实验使用Agilent 1100系统,配以四元泵、自动进样器、色谱柱恒温箱等元件。色谱柱为YMC Triart二醇基HILIC柱(4.6×250mm,3μm)。色谱条件采用梯度洗脱法,其中流动相A为含有53mM甲酸的乙腈溶液(pH约为4.0~4.5),流动相B为含有60mM的甲酸铵和53mM甲酸的水溶液(pH约为3.6)。
梯度系统由流动相A和流动相B组成;梯度系统如下:0~15min,维持2%流动相A;15~60min,将流动相A从2%提高到40%;60~70min,维持40%流动相A;70~80min,将流动相A从40%降回初始状态,即2%。上述%均为体积%。梯度系统(流动相)的流速控制在200μL/min。进样量为10μL。
3)、质谱检测
将步骤2)所得的洗脱液直接进行质谱检测。
质谱实验采用QSTAR四级杆/飞行时间质谱仪,配以TurboIonSpray离子源;负离子模式检测;扫描范围为500~1000Da。去簇电压(DP)为50V;聚焦电势(FP)为200V;离子源电压为4500V;去簇电势2(DP2)为10V;气源1(GS1)为50psi;气源2为(GS2)45psi;气帘气(CUR)为40psi;温度为400℃;子离子扫描(PIS)采用的碰撞能为20V。数据采集与分析采用Analyst QS v2.0。
结果如下:
一、按上述实验方法对刀额新对虾脂质提取物(即,步骤1)的脂质粗提物)进行HILIC-TOF/MS检测。根据洗脱顺序检测到PE,PS和PC的保留时间分别为14.82,19.90和49.87分钟(图1),其中每一类磷脂均包含多个脂肪酸链和饱和度不同的磷脂分子。复杂的磷脂分子成分对色谱峰的分辨率与峰形产生了一定影响,尤其是含量最为丰富的PC。以标准品浓度范围0.5-200μg/mL进行梯度稀释进样,标准品含量为横坐标,对应的峰面积为纵坐标,以y=a+bx线性拟合,得到PE,PS和PC的标准曲线为y=1.1×107x+12658;y=2.1×107x+98919;和y=6.3×106-29680,根据由图1可得峰面积(x),经标准曲线计算得出PE,PS和PC的总含量(y)分别为:827、106和1138μg/g。
对PC、PE及PS三类磷脂中各个分子种进行质谱分析鉴定,结果如图2。
PC主要以[M+HCOO]-离子模式存在,种类较多,主要集中在m/z 800到950区域。其中丰度最大的为m/z 850.8和878.8离子,经搜索lipidview数据库鉴定分别为PC-16:0/22:6&16:1/22:5和PC-18:0/22:6&20:1/20:5。
PE分子则以[M-H]-形式存在,响应最大的为m/z 786.6,经鉴定其两条脂肪酸链sn-1/sn-2为18:2/22:6。
PS离子同样以m/z 786.6信号最强,但结构却截然不同,其sn-1/sn-2为PS-18:0/18:2。另外响应较强的还有m/z 806.6鉴定为PS-16:0/22:6。并鉴定了23个PC分子,18个PE分子和16个PS分子,共计57个磷脂分子(表1)。
表1、刀额新对虾中PC、PE和PS磷脂分子种的结构与含量
刀额新刀额新对虾磷脂含量丰富,尤其以sn-2为DHA或EPA链的磷脂含量最高的,如16:0/22:6、16:0/20:、18:0/20:5等。
二、方法学验证
以标准品浓度范围0.5~200μg/mL进行梯度稀释进样,标准品含量为横坐标,对应的峰面积为纵坐标,以y=a+bx线性拟合,三种模型的R2均大于0.99。分别以3倍和10倍信噪比(S/N)确定目标分析物的检出限(LOD)和定量限(LOQ),测得PC、PE和PS的检出限为0.17~0.28μg/mL,定量限为0.54~0.80μg/mL。对空白肌肉组织样品添加水平分别为LOQ和20倍LOQ,每个浓度设置3个样品,按拟定方法测定,计算日内和日间精密度、回收率见表2。日内精密度小于6.2%,日间精密度小于8.1%,表明精密度良好;磷脂回收率为74%~87%,满足分析测试需要。
表2、刀额新对虾中磷脂HILIC-TOF/MS检测方法的检出限、准确性、回收率等参数的方法学验证
综上所述,利用本发明一种刀额新对虾中磷脂的亲水色谱-串联质谱检测方法,共测定了PC、PE及PS三类磷脂57个分子,刀额新对虾中的磷脂富含多不饱和脂肪酸链,其sn-2位置尤以DHA链和EPA链居多。该方法具有快捷便捷、且灵敏度、精密度及准确性好,与传统正相色谱相比避免了复杂的流动相体系和昂贵的正相色谱装置,可为脂质组学在水产食品学科中的发展奠定了一定基础。
验证实验1、采用本发明公认的检测精度高的正向色谱法对实施例1的样品进行分离定量,收集组分进行质谱检测,所得结果为:PE,PS和PC的总含量(y)分别为:806、98和1105μg/g;结构鉴定结果与表1一致。
对比例1-1、将实施例1步骤2)的“53mmol/L甲酸的乙腈溶液为流动相A,60mM甲酸铵和53mM甲酸的水溶液为流动相B”改成“26mmol/L甲酸的乙腈溶液为流动相A,30mM甲酸铵和26mM甲酸的水溶液为流动相B”;其余等同于实施例1。所得结果为PE,PS和PC的总含量(y)分别为:613、58和705μg/g。
部分的主要磷脂成分含量如下表3,其含量均低于本发明方法所对应的含量。
表3
对比例1-2、
将实施例1步骤2)的“53mmol/L甲酸的乙腈溶液为流动相A,60mM甲酸铵和53mM甲酸的水溶液为流动相B”改成“纯乙腈溶液为流动相A,纯水为流动相B”;其余等同于实施例1。所得结果为PE,PS和PC的总含量(y)分别为:419、37和545μg/g。
部分的主要磷脂成分含量如下表4,含量均低于本发明方法中的含量。
表4
对比例2、
将实施例1步骤2)的梯度系统由“0~15min,维持2%流动相A;15~60min,将流动相A从2%提高到40%;60~70min,维持40%流动相A;70~80min,将流动相A从40%降回初始状态的2%”改成“20%流动相B等度系统”;其余等同于实施例1。所得结果为PE,PS和PC的总含量(y)分别为:533、46和629μg/g。磷脂结构鉴定结果基本同表1。
对比例3、
将实施例1步骤2)的流速由“200μL/min”改成“400μL/min”;其余等同于实施例1。所得结果为PE,PS和PC的总含量(y)分别为:501、41和608μg/g。磷脂结构鉴定结果基本同表1。
对比例4、将实施例1步骤3)中的“去簇电压(DP)为50V;温度为400℃;子离子扫描(PIS)采用的碰撞能为20V”改成“去簇电压(DP)为40V;温度为400℃;子离子扫描(PIS)采用的碰撞能为10V”;其余等同于实施例1。所得结果为PE,PS和PC的总含量(y)分别为:347、17和425μg/g。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!