一种以双环铂为有效成分的药物的检测方法与流程
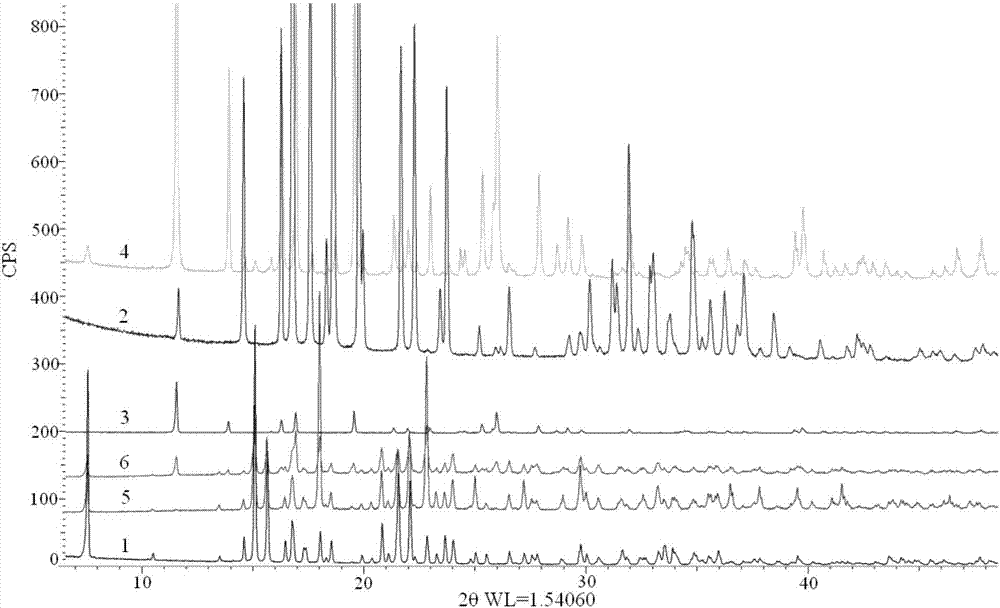
本申请是向中国知识产权局提交的申请号为201410397103.6,申请日为2014年08月13日,发明创造名称为“一种以双环铂为有效成分的药物的检测方法”的发明专利申请的分案申请。本发明涉及一种药物检测方法,特别涉及一种以双环铂为有效成分的药物的检测方法。
背景技术:
:双环铂(dicycloplatin)是由卡铂与环丁烷二酸通过四个氢键组成的相对稳定的超分子化合物,其化学结构为1,1-环丁烷二羧酸,二氨配铂(ⅱ)合1,1-环丁烷二羧酸(bis-[1,1-cyclobutanedicarboxylicaciddiaminoplatin(ⅱ)]complex)。研究表明双环铂具有广谱、高效、低毒、低耐药、低交叉耐药以及穿透性好等特点,是新一代超分子抗癌药物。例如,公开号为cn1311183a的发明专利对双环铂进行了急毒、药效实验及临床检验,结果表明双环铂不仅低毒而且抗癌活性高,对泌生癌、头颈癌、鼻咽癌、乳腺癌、肺癌、肝癌、胰腺癌、胃癌、肠癌、淋巴癌等均有显著疗效。目前,尚无对双环铂进行有效定性和/或定量的相关方法报道。技术实现要素:本发明提供一种以双环铂为有效成分的药物的检测方法,用于对双环铂进行有效的定性和/或定量分析。双环铂作为新一代超分子抗癌药物的临床功效的证明为其推广应用提供了基础,为实现该药物的工业化生产,建立相关的质量检测和监控体系不可或缺。双环铂的超分子结构来自分子中卡铂与环丁烷二酸之间的氢键结合,本发明人经研究发现,由于双环铂的这种超分子结构特性,采用液相色谱-质谱联用法(lc-ms)、流动注射-质谱分析、红外光谱分析、毛细管电泳(cze)等多数测试方法均无法对以双环铂为有效成分的药物中的双环铂进行精确地定性,尤其是无法将双环铂的化合物与卡铂和环丁二酸的物理混合物区分开,测定结果容易出现假象。因此,本发明提供一种以双环铂为有效成分的药物的检测方法,所述药物为双环铂原料药,所述检测方法包括:对双环铂原料药进行x射线粉末衍射分析,获得第一衍射图谱;确认所述第一衍射图谱中2θ角为10.3-10.7°处和/或2θ角为11.4-11.7°处是否具有衍射峰;若所述第一衍射图谱中2θ角为10.3-10.7°处具有衍射峰和2θ角为11.4-11.7°处不具有衍射峰中的一种或两种情况存在,则所述药物中含有双环铂。本发明人发现,采用x射线粉末衍射分析(xrpd)能够有效地对双环铂进行定性分析。其中,室温以透射模式得到的衍射图谱中,2θ角为10.3-10.7°处(10.51°左右处)具有衍射峰可以表明双环铂存在,而环丁二酸、卡铂、环丁二酸和卡铂的物理混合物在该处均不具有衍射峰;此外,2θ角为11.4-11.7°处(11.55°左右处)不具有衍射峰也可以表明双环铂存在,环丁二酸、卡铂、环丁二酸和卡铂的物理混合物在该处均具有衍射峰。因此,x射线粉末衍射分析尤其能够对粉末状的含有双环铂的药物进行定性检测,例如双环铂原料药。对于含有双环铂的溶液(例如双环铂针剂),本发明人发现多数的检测条件(例如液相色谱分析等)会破坏双环铂分子内氢键,使其无法以超分子的形式存在,即双环铂在溶液中完全解离成卡铂和环丁二酸,因此无法有效地对双环铂溶液中的双环铂进行定性检测,例如无法将其与卡铂和环丁二酸的混合溶液区分开。本发明人经研究发现,尽管双环铂在溶液状态下(例如水溶液)中完全解离成卡铂和环丁二酸,然而该溶液在经冻干后仅存在少量的卡铂和环丁二酸,在对双环铂溶液的水溶冻干粉末进行x射线粉末衍射分析所得到的衍射图谱中,在2θ角为10.3-10.7°处具有衍射峰和/或在2θ角为11.4-11.7°处不具有衍射峰(下称特征峰),即双环铂溶液的水溶冻干粉末与双环铂原料药具有相同的特征峰;而环丁二酸和卡铂物理混合物的水溶冻干粉中虽然形成少量双环铂,但在上述特征峰处呈现不同,即在在2θ角为10.3-10.7°处不具有衍射峰和/或在2θ角为11.4-11.7°处具有衍射峰。因此,利用xrpd能够有效地对双环铂溶液中的双环铂进行定性检测,从而将其与卡铂和环丁二酸的混合溶液区分开。因此,本发明还提供一种以双环铂为有效成分的药物的检测方法,所述药物为双环铂针剂,所述检测方法包括:对双环铂针剂进行冻干处理,制得双环铂药粉;对所述双环铂药粉进行x射线粉末衍射分析,获得第一衍射图谱;确认所述第一衍射图谱中2θ角为10.3-10.7°处和/或2θ角为11.4-11.7°处是否具有衍射峰;若所述第一衍射图谱中2θ角为10.3-10.7°处具有衍射峰和2θ角为11.4-11.7°处不具有衍射峰中的一种或两种情况存在,则所述药物中含有双环铂。在本发明中,所述药物中含有双环铂,其为该药物的有效成分;此外,该药物中可能还含有未结合成双环铂或者由双环铂解离所形成的卡铂和/或环丁二酸,本发明将这些卡铂和/或环丁二酸定义为杂质。具体地,本发明可以通过三种方式对药物中的双环铂进行定性检测,即:1)若所述第一衍射图谱中2θ角为10.3-10.7°处具有衍射峰,则所述药物中含有双环铂;2)若所述第一衍射图谱中2θ角为11.4-11.7°处不具有衍射峰,则所述药物中含有双环铂;3)若所述第一衍射图谱中2θ角为10.3-10.7°处具有衍射峰和2θ角为11.4-11.7°处不具有衍射峰,则所述药物中含有双环铂。并且,上述第2)和第3)种情况说明该药物中不含有所述杂质。进一步地,本发明人发现在以双环铂为有效成分的药物,所述杂质(尤其是卡铂)表现为在2θ角为11.4-11.7°处具有衍射峰。因此,本发明还提供了对药物中所述杂质进行定量检测的方法,即:若所述第一衍射图谱中2θ角为10.3-10.7°处和2θ角为11.4-11.7°处均具有衍射峰,则所述检测方法还包括:将双环铂标准品与杂质制成混合粉末进行x射线粉末衍射分析,获得第二衍射图谱,所述混合粉末中杂质的质量百分含量为x%;分别获得所述第一衍射图谱中2θ角为10.3-10.7°处的衍射峰的积分强度a11和2θ角为11.4-11.7°处的衍射峰的积分强度a12,以及分别获得所述第二衍射图谱中2θ角为10.3-10.7°处的衍射峰的积分强度a21和2θ角为11.4-11.7°处的衍射峰的积分强度a22,计算a12/a11和a22/a21;若a12/a11≤a22/a21,则所述药物中杂质的含量≤x%,若a12/a11>a22/a21,则所述药物中杂质的含量>x%;其中,所述杂质为卡铂和环丁二酸中的一种或两种。在本发明中,可以药物中的卡铂含量(杂质为卡铂)作为药物杂质的评价指标来对药物中的杂质含量进行评价,特别是所述x%可以为0.1-1%。此时,若a12/a11≤0.67,则所述药物中卡铂的含量≤1%;若a12/a11>0.67,则所述药物中卡铂的含量>1%;进一步地,若a12/a11≤0.55,则所述药物中卡铂的含量≤0.5%;若a12/a11>0.55,则所述药物中卡铂的含量>0.5%;再进一步地,若a12/a11≤0.5,则所述药物中卡铂的含量≤0.1%;若a12/a11>0.5,则所述药物中卡铂的含量>0.1%。在本发明中,所述药物中卡铂含量的检测限可达0.1%。在本发明具体方案中,采用配有透射旋转样品台的x射线粉末衍射仪进行所述x射线粉末衍射分析,方法包括:使用单色cukα辐射,管压为40kv,管流为40ma,2θ扫描范围为6-55°,扫描速度为0.6s/步,步长为0.015°/步。所述第一衍射图谱和第二衍射图谱均是在室温以透射模式得到。进一步地,本发明的检测方法还包括对以双环铂为有效成分的药物进行定量检测,即:将双环铂原料药制成溶液后或双环铂针剂稀释后进行反相高效液相色谱检测,同时以0.025-0.999mg/ml的卡铂溶液和0.101-3.99mg/ml的环丁二酸溶液作为标准溶液,分别确定所述双环铂针剂中卡铂和环丁二酸的质量含量。进一步地,使所述溶液或稀释后的所述双环铂针剂中双环铂的质量浓度为1mg/ml,并且以0.7mg/ml的卡铂溶液和0.3mg/ml的环丁二酸溶液作为标准溶液。所述质量含量的确定可以通过外标法进行。本发明还可以采用其它定量检测方法,例如:将双环铂原料药制成溶液后或双环铂针剂稀释后进行反相高效液相色谱检测,同时以0.067-2.7mmol/l的卡铂溶液和0.7-27.8mmol/l的环丁二酸溶液作为标准溶液,分别确定所述双环铂针剂中卡铂和环丁二酸的摩尔含量以及卡铂与环丁二酸的摩尔比。进一步地,使所述溶液或稀释后的所述双环铂针剂中双环铂的摩尔浓度为2mmol/l,并且以2mmol/l的卡铂溶液和2mmol/l的环丁二酸溶液作为标准溶液。在本发明具体方法中,所述反相高效液相色谱的条件为:色谱柱:aq-c18,4.6×250mm,5μm;检测波长:220nm;进样量:10μl;流速:1.0ml/min;流动相:a相为20mmol/l且ph为3.0的磷酸二氢钠缓冲液,b相为甲醇;梯度条件:0-3min,5%b相,3-8min,5-60%b相,8-12min,60%b相。本发明所提供的以双环铂为有效成分的药物的检测方法,能够对药物中的双环铂进行定性和/或定量分析,其能够排除其它测定方法所带来的假象,真实且直观地对双环铂进行有效地测定,尤其是本发明能够将其与卡铂与环丁二酸的物理混合物区分开,并且能够对药物中的杂质含量(以卡铂计)进行有效地测定,从而为双环铂药物的质量控制奠定了基础。附图说明图1为本发明实施例1中样品1在透射模式和反射模式下的x射线粉末衍射图谱,其中a为透射模式,b为反射模式;图2为本发明实施例1各样品的x射线粉末衍射图谱;图3为本发明实施例2各样品的x射线粉末衍射图谱;图4为本发明实施例4各样品的dsc曲线;图5为本发明对照例1的样品1的红外图谱;图6为本发明对照例1的样品4的红外图谱;图7为本发明对照例2的双环铂针剂在正离子模式下的质谱图;图8为本发明对照例2的双环铂针剂在负离子模式下的质谱图。具体实施方式为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的附图和实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明实施例所采用的样品包括:双环铂原料药(以下称样品1):四批,均购自北京市兴大科学系统公司,批号分别为:20130115-1、20130115-2、2013015-3和20130216;环丁二酸(以下称样品2):纯度为99.5%以上;卡铂(以下称样品3):纯度为99%以上;环丁二酸和卡铂的物理混合物(以下称样品4):取19mg环丁二酸和50mg卡铂,混合后于研钵中轻微研成粉末状,其中环丁二酸与卡铂的摩尔比约为1:1;在本发明中,将环丁二酸和卡铂直接混合所得到的混合物定义为环丁二酸和卡铂的物理混合物,该混合物中环丁二酸和卡铂未形成氢键;双环铂原料药的水溶冻干粉(以下称样品5):取60mg双环铂原料药,用2.5ml去离子水溶解,室温放置2h后,置于-70℃冰箱冷冻4h,转移至冷冻干燥机(冷阱温度-69℃,真空为10pa),冷冻干燥12h,制得白色冻干粉;环丁二酸和卡铂物理混合物的水溶冻干粉(以下称样品6):将上述环丁二酸和卡铂的物理混合物按照上述方法制成冻干粉;双环铂标准品和针剂:购自北京市兴大科学系统公司,其中:双环铂针剂为5ml安瓿,双环铂的含量为1%(即1g/100ml);双环铂针剂的冻干粉:将上述双环铂针剂按照上述方法制成冻干粉。实施例1x射线粉末衍射(xrpd)分析一、仪器和条件x射线粉末衍射仪:brukerd8-advance,配有反射和透射旋转样品台;透射:cukα辐射,聚焦单色器,gobel-mirror聚焦光路,管压40kv管流40ma;扫描方式:θ/2θ扫描;ds发散狭缝1.2mm,索拉狭缝2.5mm;2θ扫描范围:6-55°;扫描速度:0.6s/step;步长:0.015°/step。二、xrpd分析在室温下采用透射模式和反射模式分别对一批样品1进行xrpd测定,衍射图谱如图1所示。由图1可知:鉴于双环铂原料药为针状晶体,存在严重的择优取向,相对于反射模式(b),采用透射模式(a)能够弱化双环铂的择优取向,从而排除晶体形状干扰,因此能够更为真实地反映样品的结构信息。在本实施例中,xrpd均在室温下采用透射模式进行。将上述各样品分别装入透射样品台后进行xrpd测定,衍射图谱如图2所示。由图2可知:含有双环铂的样品1和样品5与其它样品存在显著差异,首先,样品1和5在2θ角为11.4-11.7°处(11.55°左右)无明显的衍射峰,而其它几个样品在该处具有明显的衍射峰;其次,样品1和5在2θ角为10.3-10.7°处(10.51°左右)具有明显的衍射峰,而其它样品在该处无明显的衍射峰。因此,可以通过衍射图谱中2θ角为10.3-10.7°处和/或2θ角为11.4-11.7°处是否具有衍射峰来判定样品中是否含有双环铂,从而将其与卡铂、环丁二酸、环丁二酸和卡铂的物理混合物及其水溶冻干粉区分开。进一步地,对上述双环铂针剂的冻干粉进行xrpd测定,其衍射图谱与样品5基本相同,衍射图谱中在2θ角为10.3-10.7°处具有衍射峰,而在2θ角为11.4-11.7°处不具有衍射峰。实施例2药物杂质分析精密称取4份50mg双环铂标准品(不含卡铂和环丁二酸),向各份分别添加0.5mg、0.25mg、0.05mg和0mg卡铂,轻微研细混合均匀,分别制成样品a、样品b、样品c、样品d,各样品的卡铂含量分别为1%、0.5%、0.1%和0%。将上述样品a-d分别装入透射样品台后,按照实施例1方法进行xrpd测定,衍射图谱如图3所示。将衍射图谱中2θ角为10.3-10.7°处的衍射峰的积分强度记作a21,2θ角为11.4-11.7°处的衍射峰的积分强度记作a22。经计算,样品a-c的a22/a21分别为0.67、0.55和0.5。对双环铂原料药进行xrpd测定,并将其衍射图谱中2θ角为10.3-10.7°处的衍射峰的积分强度记作a11,2θ角为11.4-11.7°处的衍射峰的积分强度记作a12,计算a12/a11,并通过各样品的卡铂含量及其相对应的a22/a21作为参照来确定双环铂原料药中卡铂的含量。即,若a12/a11≤0.67,则双环铂原料药中卡铂的含量≤1%,若a12/a11>0.67,则双环铂原料药中卡铂的含量>1%;进一步地,若a12/a11≤0.55,则双环铂原料药中卡铂的含量≤0.5%,若a12/a11>0.55,则双环铂原料药中卡铂的含量>0.5%;再进一步地,若a12/a11≤0.5,则双环铂原料药中卡铂的含量≤0.1%,若a12/a11>0.5,则双环铂原料药中卡铂的含量>0.1%。本实施例仅示例了如何对双环铂原料药中的卡铂含量进行定量检测,然而上述样品中的卡铂含量并不局限于上述所示例的数值,只要能够添加通过xrpd衍射图谱检测出样品中的卡铂含量(例如样品中卡铂含量≥0.1%),则a22/a21可以是其它任意数值,并且可以该数值作为参照来确定双环铂原料药中的卡铂含量。实施例3反相高效液相色谱分析一、仪器和色谱条件仪器:岛津lc-20a高效液相色谱系统,配备spd-10a紫外-可见检测器,自动进样器,lcsolution化学工作站;色谱柱:aq-c18,4.6×250mm,5μm;检测波长:220nm;进样量:10μl;流速:1.0ml/min;流动相及梯度条件:a相:20mmol/l磷酸二氢钠缓冲液,ph3.0;b相:甲醇;梯度条件:0-3min,5%b相,3-8min,5-60%b相,8-12min,60%b相。二、卡铂和环丁二酸的线性关系分别精确称取卡铂9.99mg、环丁二酸39.94mg,置10ml容量瓶中,用流动相溶解并定容至刻度,制成0.999mg/ml的卡铂溶液和3.99mg/ml的环丁二酸溶液;经逐级稀释后,制得一系列不同浓度的溶液,然后分别取10μl溶液注入液相色谱仪进行测定。液相色谱结果表明:卡铂溶液在0.025-0.999mg/ml(即0.067-2.7mmol/l)范围内与峰面积积分值呈现良好的线性关系,线性回归方程的相关系数r为0.999;环丁二酸溶液在0.101-3.99mg/ml(即0.7-27.8mmol/l)范围内与峰面积积分值呈现良好的线性关系,线性回归方程的相关系数r为0.999。三、双环铂药物的定量测定分别精密称取上述四批双环铂原料药各约10mg至10ml容量瓶,用流动相溶解,配制成约1.0mg/ml的溶液;同时对上述双环铂针剂进行稀释,制成约1.0mg/ml的溶液。分别取10μl上述各溶液注入液相色谱仪进行测定;同时以0.7mg/ml左右的卡铂溶液和0.3mg/ml左右的环丁二酸溶液作为标准溶液,采用外标法进行定量测定后,计算测定结果相对于各自标准溶液的质量百分含量(即,测定的卡铂或环丁二酸的浓度/标准溶液的浓度×100%),结果如表1所示。表1双环铂药物中卡铂和环丁二酸的质量百分含量双环铂药物卡铂的质量百分含量环丁二酸的质量百分含量第一批原料药101.8%102.7%第二批原料药100.0%100.4%第三批原料药98.3%100.0%第四批原料药101.3%102.6%双环铂针剂99.8%97.6%由表1结果可知:双环铂药物中卡铂和环丁二酸的含量在97-103%,其可作为双环铂药物的质量控制指标之一。进一步地,还可以采用下述方法进行定量测定:将上述四批双环铂原料药,分别配制成摩尔浓度约为2mmol/l的溶液;同时将上上述双环铂针剂进行稀释为2mmol/l左右的溶液。分别取10μl上述各溶液注入液相色谱仪进行测定;同时以2mmol/l左右的卡铂溶液和2mmol/l左右的环丁二酸溶液作为标准溶液,采用外标法进行定量,结果如表2所示。表2双环铂药物中卡铂和环丁二酸的摩尔浓度及摩尔比由表2结果可知:双环铂药物中卡铂与环丁二酸的摩尔比为0.95-1.05,其可作为双环铂药物的质量控制指标之一。实施例4示差扫描量热法分析(dsc)一、仪器和参数示差扫描量热仪:美国taq2000,al盘为参比盘,样品盘为铝盘,升温速率:10℃/min,升温区间40℃-240℃。二、示差扫描量热法分析分别精确称取50mg样品1至样品6,并用示差扫描量热仪测定其热重参数,结果如图4所示。图4结果表明:样品1、样品5和样品6的dsc曲线较为相似,其分别在197.82℃、198.23℃和184.13℃开始出现吸热峰;样品4与样品1的dsc曲线呈现较大差异,其除在182.04℃处有吸热峰外,在153.05℃还有一个明显的吸热峰,该峰与环丁二酸的吸热峰接近,可确认为环丁二酸组分吸热峰;而样品3在153℃和200℃附近均未出现吸热峰,当温度升至252℃时有一个分解峰。此外,样品1和样品5在197℃出现尖峭的相变峰,吸收峰仅略发生位移,说明冻干过程对双环铂两个组分间的影响不大;样品6与样品1和样品5的dsc曲线相似,其在153℃附近未出现环丁二酸熔融峰,说明样品6中卡铂与环丁二酸由于氢键作用形成了双环铂类似物,然而其在197℃附近的峰形很宽,由此说明双环铂的两个组分处于结晶态,而卡铂和环丁二酸物理混合物的水溶冻干粉的组分处于半结晶态或无定形态,其结晶状态与双环铂显著不同。由此说明,双环铂原料药与卡铂和环丁二酸的物理混合物存在显著差异,双环铂的两个组分间由于氢键作用而形成了新的超分子氢键簇集体。实施例5核磁共振滴定法(nmr)一、仪器和参数核磁共振滴定采用日本电子eca-400型超导傅立叶变换核磁共振仪,其配有选择脉冲laminal波形发生器和5mmz-轴梯度脉冲多核探头。1h工作频率分别为400mhz,以dmso-d6为溶剂,tms为内标;实验温度为室温,使用ф5mm多核探头;1hnmr的谱宽9.18khz,数据点32768,90°脉冲宽度11μs,弛豫延迟1.2s。二、核磁共振滴定实验(固定环丁二酸量)固定环丁二酸的用量并改变卡铂的加入量,用0.5ml氘代dmso溶解后放置一夜,测定环丁二酸羧基与卡铂氨基的1hnmr化学位移,卡铂、环丁二酸的添加量与核磁滴定结果见表3。表3结果表明,除环丁二酸分子中的羧基氢外,其它所有氢原子的化学位移均未随卡铂加入量的增加发生改变,说明卡铂分子中的氨基氢受环丁二酸的加入影响较小。并且,环丁二酸羧基氢的化学位移随其自身加入量的增加而发生显著变化,从高场往低场发生移动(12.68→13.42ppm),由此说明环丁二酸与卡铂之间的确存在氢键作用。此外,依据不同卡铂添加量下的羧基氢的化学位移值(p),通过非线性拟合计算得卡铂与环丁二酸之间的解离常数为0.3mmol/l;依据羧基氢的化学位移值倒数与卡铂摩尔浓度的倒数,计算得环丁二酸与卡铂的结合常数为4.22x104l/mol;由此进一步说明双环铂的两个组分之间确实存在氢键作用,但两个组分之间的氢键结合力较弱。表3卡铂和环丁二酸的添加量及化学位移对照例1红外光谱分析采用perkinelmer红外光谱仪(frontier)对样品1和样品4进行测定,测定方法具体为:预先将kbr烘干,研磨,压片后测本底,再将样品1、样品4分别与适量kbr粉末研磨混匀,压片后测定,样品1和样品4的红外图谱分别如图5和图6所示。图5和图6结果表明:样品1和样品4的红外图谱无显著差异,表明红外光谱分析无法对双环铂原料药与卡铂和环丁二酸的物理混合物进行有效区分,因此无法对双环铂进行有效且精确地定性分析。对照例2液相色谱-质谱联用(lc-ms)一、仪器及色谱条件毛细管电压:3kv(或-2.6kv);提取锥孔电压:4v(或-4v);样品锥孔电压:15v(或-20v);源温:300℃;扫描范围:80-1000;色谱柱:xdb-c18,4.6x50mm,1.8um;流动相及梯度条件:流动相a:0.1%甲酸水溶液;流动相b:甲醇;梯度:0min,1%b;1min,1%b;3.5min,60%b;5.5min,60%b;流速:0.5ml/min;进样体积:2ul。二、液相色谱-质谱分析按照上述色谱条件对双环铂针剂进行液相色谱-质谱分析,结果如图7和图8所示。图7和图8结果表明:双环铂在液相色谱分离条件下无法以其超分子氢键簇集体形式存在,其完全解离成卡铂与环丁二酸,其中在正离子和负离子模式下分别检测到解离产生的卡铂和环丁二酸,图7中m/z372.0514为卡铂的分子离子峰[m+h]+,图8中m/z143.0341为环丁二酸的分子离子峰[m-h]-。由此表明:lc-ms同样无法对双环铂溶液进行有效地定性分析。对照例3流动注射-质谱分析鉴于液相色谱分离会破坏双环铂分子内的氢键,因此尝试采用流动注射进样,从而直接对双环铂水溶液(双环铂针剂)进行分析。结果表明:双环铂水溶液在正离子模式下仅检测到卡铂的分子离子峰[372.0543],而在负离子模式下观察到双环铂[m/z514.0917]和环丁二酸[m/z143.0375]的分子离子峰。由此可见,色谱分离会破坏双环铂超分子氢键簇集体,因此无法区分双环铂水溶液与卡铂和环丁二酸的混合溶液;采用流动注射进样后直接质谱分析能够在负离子模式下获得双环铂的准确分子量,因此其仅适用于对双环铂的准确分子量进行测定,不适合对双环铂水溶液中的双环铂进行定性检测。对照例4毛细管电泳(cze)一、仪器和参数采用agilentg1602a毛细管电泳仪;电压:+20kv;毛细管内径:75μm;紫外/二极管阵列监测器。二、水相cze分离配制四份20mm的ph为7.05左右的nah2po4-na2hpo4缓冲液,向其中三份缓冲液中分别添加β-环糊精(β-cd)、羟丙基-β-环糊精(hp-β-cd)和羧甲基-β-环糊精(cm-β-cd),该四种缓冲液作为毛细管电泳分离缓冲液;开启电泳仪后,分别用1m的naoh、0.1m的naoh、水、上述四种缓冲液依次冲洗毛细管柱,进样后在电压为20kv、柱温为25℃下对样品1至样品4进行分离。结果表明,在上述各分离条件下(即各缓冲液),双环铂与卡铂的出峰时间均一致,未发现有分离迹象,卡铂、双环铂、卡铂和双环铂的物理混合物的出峰时间均在7.42min左右,并且双环铂与卡铂的物理混合物的峰面积约为卡铂和双环铂的峰面积之和。三、非水cze分离(nace)配制20mmnh4ac的乙腈溶液,用乙酸调节ph至7左右后作为毛细管电泳分离缓冲液,按照上述方法对样品1至样品4进行分离。结果表明,在上述分离条件下,双环铂与卡铂未发现有分离迹象。四、胶束毛细管电泳分离(mekc)配制20mm的十二烷基磺酸钠(sds)水溶液作为毛细管工作缓冲液,按照上述方法对样品1至样品4进行分离。结果表明,在上述分离条件下,双环铂与卡铂未发现有分离迹象。综上所述,采用三种模式的毛细管电泳对各样品进行分离时,各样品均未出现分离现象,且双环铂和卡铂的出峰时间一致,其进一步说明双环铂由于超分子内相对共价键较弱的氢键存在,在电场能量环境作用下解离为卡铂和环丁二酸。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1