蓝花药中主要活性成分的HPLC检测方法与流程
本发明属于医药技术领域,涉及中成药中活性成分的检测方法,具体的说是一种同时测定蓝花药中三种活性成分的hplc检测方法。
背景技术:
蓝花药收载于部颁标准中药成方制剂第三册(ws3-b-0647-91),具有清热解毒,消痈等功效,用于治疗急慢性扁桃腺炎、齿龈红肿、咽喉肿痛,小儿疳症。蓝花药的处方为硼砂100g;冰片45g;青黛60g;玄明粉60g;滑石粉50g;琥珀;50g;枯矾20g;石膏150g;儿茶30g;胡黄连30g。蓝花药的制法为以上十味,除冰片外,其余硼砂等九味粉碎成最细粉,将冰片研细,与上述粉末配研,过筛,混匀,用水泛成极小丸。另取滑石粉,药用炭等量混合,制成桂衣粉,用蔗糖10g,桃胶5g加水制成45g稀胶糖浆作为粘合剂。将水丸包衣(每300g干丸用挂衣粉及粘合剂各27g),打光低温干燥,即得。处方中的每种药材虽然每种成分的含量不同,但是都具有重要的药理协调作用。蓝花药原质量标准只进行了青黛、硼砂、冰片的理化鉴别,专属性不强。张亚秋等人在《时珍国医国药》第2006年第17卷第5期所公开的“蓝花药的质量标准研究”,对该药物的质量控制方法作了补充,增加了处方中青黛、冰片、琥珀、滑石粉和石膏的显微鉴别、青黛和冰片的薄层分析鉴别及硼砂含量测定项。中国专利(cn101390984b)公开了“蓝花药的检测方法”,提供了一种用高效液相色谱法测定青黛含量、用气相色谱法测定冰片含量以及用薄层色谱法对冰片、儿茶进行定性鉴别。然而,目前尚未见利用高效液相色谱法同时测定蓝花药中多种活性成分。因此必需建立一种快速、准确、全面分离、检测蓝花药多种活性成分的方法,以便更好地对该产品进行质量监控和用药安全。
技术实现要素:
本发明目的在于提供一种利用高效液相色谱法同时测定蓝花药中儿茶的儿茶素与表儿茶素;胡黄连中胡黄连苷ⅰ的含量,为判断该药的质量标准提供理论依据。该方法简单、稳定、重现性好,可用于该药品的质量控制。
本发明的目的是这样实现的:该测定方法是利用高效液相色谱法(hplc)在同一实验条件下同时分离并测定蓝花药中儿茶素、表儿茶素和胡黄连苷ⅰ的含量,具体步骤如下:
(1)混合对照品溶液的制备:准确称取儿茶素3.61mg、表儿茶素0.83mg、胡黄连苷ⅰ0.35mg置于10ml容量瓶中,加入5ml甲醇溶解后,定容,制成混合对照品溶液。儿茶素、表儿茶素和胡黄连苷ⅰ的浓度分别为0.3610mg·ml-1、0.0830mg·ml-1和0.0350mg·ml-1。
(2)样品溶液的制备:取蓝花药样品,研细,混匀,精确称6.5123g,放入锥形瓶中,精密加入50ml甲醇,称定重量,超声处理1h,放冷,补足减失的重量,混匀,离心10min(转速为4000转),取上层清液,滤过,即得。
(3)阴性样品溶液的制备:按照处方工艺去除儿茶、胡黄连制得样品,取制备的阴性样品,研细,混匀,精确称6.4120g,放入锥形瓶中,精密加入50ml甲醇,称定重量,超声处理1h,放冷,补足减失的重量,混匀,离心10min(转速为4000转),取上层清液,滤过,即得。
(4)分离检测:色谱柱:tc-c18(4.6mm×250mm,5μm);流动相:甲醇:0.5%醋酸梯度洗脱,采用梯度洗脱方式为:
流速:1.0ml·min-1;检测波长:278nm;柱温:25℃;进样量:10μl。
本发明具有以下优点和积极效果:
1、本发明方法采用高效液相色谱法,利用梯度洗脱方式,实现了同时测定蓝花药中三种有效成分的含量;该色谱法能够较好的分离、测定蓝花药中儿茶素、表儿茶素和胡黄连苷ⅰ有效成分的含量,且色谱峰峰形好,分离度较高,在此色谱条件下蓝花药中儿茶素、表儿茶素和胡黄连苷ⅰ的浓度与峰面积呈现良好的线性关系。
2、本发明所述方法简单、准确、精密度、重现性和稳定性好,可为全面评价和控制蓝花药质量奠定了理论基础和科学依据。
附图说明
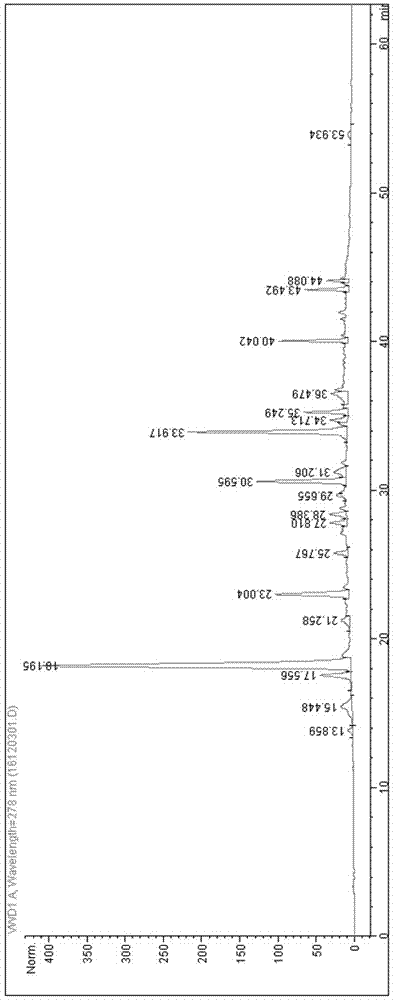
图1本发明混合对照品溶液的色谱图。
图2本发明样品溶液的色谱图。
图3本发明阴性样品溶液的色谱图。
图4本发明对比例1条件下的色谱图。
图5本发明对比例2条件下的色谱图。
图6本发明儿茶素标准曲线。
图7本发明表儿茶素标准曲线。
图8本发明胡黄连苷ⅰ标准曲线。
具体实施方式
以下结合附图和具体实施方式对本发明作以进一步的阐述,以便本领域的技术人员更了解本发明,但并不以此限制本发明。
仪器与试药
仪器:安捷伦1100高效液相色谱仪(agilent科技有限公司);bt-125d十万分之一电子天平(赛多利斯科学仪器(北京)有限公司);gwa-un4系列超纯水器(北京普析通用仪器有限责任公司);sk5200h超声波清洗器(200w59hz上海科导仪器有限公司);shz-d型循环水真空泵(上海华光仪器仪表厂);高速万能粉碎机(天津市泰斯特仪器有限公司);yxj-2台式高速离心机(江苏金坛市亿通电子有限公司)。
试药:蓝花药样品(按照处方工艺制成三批,批号为20160521;20160612;20160917);蓝花药阴性样品(按照处方工艺去除儿茶、胡黄连制成);蓝花药儿茶素对照品(中国食品药品研究院,批号:877-200001);表儿茶素对照品(中国食品药品研究院,批号:110878-200102);胡黄连甘ⅰ对照品(中国食品药品研究院,批号:111727-200501);甲醇(tedia,ms1992-801);乙腈(tedia,as1122-801);醋酸(天津市福晨化学试剂厂,批号:20090207);水为超纯水。
实施例1
(1)混合对照品溶液的制备:准确称取儿茶素3.61mg、表儿茶素0.83mg、胡黄连苷ⅰ0.35mg置于10ml容量瓶中,加入5ml甲醇溶解后,定容,制成混合对照品溶液。儿茶素、表儿茶素和胡黄连苷ⅰ的浓度分别为0.3610mg·ml-1、0.0830mg·ml-1和0.0350mg·ml-1。
(2)样品溶液的制备:取批号为20160917样品,研细,混匀,精确称6.5123g,放入锥形瓶中,精密加入50ml甲醇,称定重量,超声处理1h,放冷,补足减失的重量,混匀,离心10min(转速为4000转/min),取上层清液,滤过,即得。
(3)阴性样品溶液的制备:取制备的阴性样品,研细,混匀,精确称6.4120g,放入锥形瓶中,精密加入50ml甲醇,称定重量,超声处理1h,放冷,补足减失的重量,混匀,离心10min(转速为4000转/min),取上层清液,滤过,即得。
(4)分离检测:色谱柱:tc-c18(4.6mm×250mm,5μm);流速:1.0ml·min-1;检测波长:278nm;柱温:25℃;进样量:10μl;流动相:甲醇:体积比为0.5%的醋酸水溶液梯度洗脱程序如下:
检测结果:此时待测组分的色谱峰能够达到很好的分离,峰形对称。上述色谱条件下,混和对照品溶液的色谱图为图1,样品溶液的色谱图为图2,阴性样品溶液的色谱图为图3。从图1可以看出,儿茶素、表儿茶素和胡黄连苷ⅰ的保留时间分别为18.179min、22.914min和33.852min。
对比例1
区别在于步骤(3)分离检测时,流动相为:甲醇:体积比为0.5%的醋酸水溶液梯度洗脱,其余步骤同实施例1,所得色谱图如图4。
检测结果:在对比例1实验条件下,虽然样品分析时间较短,但保留时间为10.138min(儿茶素)组分的色谱峰没有达到很好的分离。
对比例2
区别在于步骤(3)分离检测时,流动相为:甲醇:水溶液梯度洗脱,其余步骤同实施例1,所得色谱图如图5。
检测结果:待测组分的色谱峰主要集中在7~15min中内,没有达到很好的分离。
实施例1色谱条件的方法学考查
精密度实验:准确吸取混合对照品溶液连续进样6次,每次进样10μl,根据色谱图中的峰面积,计算儿茶素、表儿茶素和胡黄连苷ⅰ的rsd分别为1.13%、1.28%和1.25%(n=6),上述数据说明仪器精密度良好。
重现性实验:取批号为20160917样品6份,按照供试品溶液的制备方法处理样品,并依照选择的色谱条件进样测试,得出儿茶素的rsd:0.95%、表儿茶素的rsd:2.45%、胡黄连苷ⅰ的rsd:4.48%,说明该方法重复性较好。
稳定性试验:取批号为20160917样品6份,按照以上的色谱条件每隔三个小时测定一次,共测定6次,儿茶素、表儿茶素和胡黄连苷ⅰ的rsd分别为0.60%、2.74%和2.06%,实验数据显示样品溶液在15h内基本稳定。
回收率试验:取已知含量的批号为20160917样品6份,分别精密加入甲醇溶解的儿茶素、表儿茶素和胡黄连苷ⅰ对照品溶液,按照样品溶液的制备方法处理样品,每次进样10μl,测得6组数据,计算回收率,结果见表1。
表1加样回收率实验结果
线性关系及含量测定:精确称取不同儿茶素、表儿茶素和胡黄连苷ⅰ标准品分别置于10ml容量瓶中,用甲醇定容,制备不同浓度的标准溶液(如表2),分别取10μl进行分析(结果见图6-8)。以对照品浓度为横坐标,峰面积为纵坐标,作线性回归方程,得各组分回归方程结果见表3。通过回归方程,根据测得峰面积数据计算,得三批蓝花药中儿茶素;表儿茶素;胡黄连苷ⅰ的含量见表4。
表2标准溶液浓度
表3线性关系方程
表4三批样品含量
综上,本发明所建立的高效液相色谱法同时测定蓝花药的主要活性成分含量,具有操作简便、精密度好、重复性高的特点,可全面、更有效地控制该药的质量。
- 还没有人留言评论。精彩留言会获得点赞!