一种食品接触材料中环氧氯丙烷迁移量的测定方法与流程
本发明涉及环氧氯丙烷测定领域,尤其涉及一种食品接触材料中环氧氯丙烷迁移量的测定方法。
背景技术:
环氧氯丙烷(3-氯-1,2-环氧氯丙烷)是一种含氧物质的稳定剂和化学中间体,其作为一种化工原料,应用广泛。然而,由于其具有毒性及对人体的伤害,国内外主要国家都将其列为限制物质。目前,除了对水中的环氧氯丙烷残留有限制要求外,对食品接触材料中环氧氯丙烷残留也有限制,如我国gb9685-2008中限定环氧氯丙烷含量的限量为1.0mg/kg;又如欧盟eu10/2011指令要求塑料中环氧氯丙烷迁移量<0.01mg/kg,日本食品卫生法则规定金属表面涂层中的环氧氯丙烷在戊烷中的溶出量≤0.5mg/l。
目前环氧氯丙烷研究主要以水中的含量为主,对食品接触材料中迁移量的研究还很少。专利号为zl201410055719.5(授权公告号为cn103760290b)的中国发明专利公开了一种食品接触材料中环氧氯丙烷迁移量的测定方法,其包括以下步骤:步骤一,制备标准工作溶液、食品模拟物试液和空白试液;步骤二,采用气相色谱-串联三重四级杆质谱联用仪对步骤一的三种溶液分别进行气相色谱—串联质谱测定;以标准工作溶液中环氧氯丙烷的浓度x为横坐标,以对应的测得的定量离子峰面积y为纵坐标,绘制标准工作溶液回归曲线,根据曲线得到的线性方程y=ax+b,计算出回归曲线的斜率a和截距b;步骤三,根据公式c=【(y模-y空)-b】/a,计算食品模拟物试液中环氧氯丙烷浓度。该专利公开了一种采用外标法来测定食品接触材料中环氧氯丙烷迁移量的方法,虽然该专利能在一定程度上较准确地测定食品接触材料中环氧氯丙烷的迁移量,但其结果随前处理过程和色谱条件影响较大,回收率不高。
此外,目前环氧氯丙烷测定的前处理方法主要有吹扫捕集、固相萃取、液液萃取等,其中,吹扫捕集法仪器配置要求高、而且检出限高;固相萃取和液液萃取方法萃取过程容易造成组分损失;采用同位素稀释-气质联用法是选用同位素稀释剂作为内标物消除检测过程误差的一种方法,但同位素稀释剂一般价格不菲。
技术实现要素:
本发明所要解决的技术问题是针对现有技术而提供一种测定结果准确性高的食品接触材料中环氧氯丙烷迁移量的测定方法。
本发明解决上述技术问题所采用的技术方案为:一种食品接触材料中环氧氯丙烷迁移量的检测方法,其特征在于包括以下步骤:
(1)标准溶液配制:分别配制一定浓度的环氧己烷标准中间溶液和环氧氯丙烷标准中间溶液;取适量的上述环氧氯丙烷标准中间溶液,加入一定量的环氧己烷标准中间溶液,定容配制得到至少三种浓度的标准工作液;
(2)样品前处理:通过食品接触材料与水基食品模拟物的迁移实验获得模拟液,然后加入一定量的上述环氧己烷标准中间溶液作内标,倒入分液漏斗中,加入氯化钠,充分溶解后加入二氯甲烷,静置,取二氯甲烷萃取液至顶空瓶;再重复上述实验步骤2次,二氯甲烷萃取液可适当减少,收集至同一顶空瓶中;
(3)气相色谱测定:采用气相色谱法对上述步骤(1)和步骤(2)获得的标准工作液和萃取液分别进行测定,按下列公式(1)计算获得模拟液中的环氧氯丙烷浓度,
式(1)中:
x——模拟液中环氧氯丙烷的含量,单位为mg/l;
ai——萃取液中环氧氯丙烷的峰面积;
ci——标准工作液中环氧氯丙烷的含量,单位为mg/l;
vi——二氯甲烷体积,单位为ml;
asc——标准工作液中环氧己烷的峰面积;
as——标准工作液中环氧氯丙烷峰面积;
v——模拟液体积,单位为ml;
ass——萃取液中环氧己烷的峰面积,其中vi按等量环氧己烷标准中间溶液定容体积10ml来计算。
作为优选,所述步骤(2)中样品前处理的具体方法为:准确移取一定体积的经迁移试验得到的模拟液,放入分液漏斗中,加入体积为上述模拟液0.5~1.5‰的环氧己烷标准中间溶液,加入氯化钠,该氯化钠与上述模拟液的质量比为4~5:100,振摇溶解,加入体积为上述模拟液4~5%的二氯甲烷,振摇1~2min;静置分层,放出下层二氯甲烷萃取液,置于顶空瓶中,盖塞,按照上述步骤,再分别准确加入二氯甲烷2~3ml和1~2ml,萃取模拟物,合并萃取液于上述顶空瓶中,从而获得所需的萃取液。
作为优选,所述步骤(3)中气相色谱测定的条件为:色谱柱:hp-innowax,柱温:40℃恒定0.5min,以20℃/min升温速率升温至105℃,恒定4.00min,以40℃/min升温速率升温至200℃,恒定1.00min;气化温度:105℃;载气为n2;其流速及进样方式:恒流,4.0ml/min,不分流;自动进样器进样,进样量1.0μl;fid检测器:温度200℃;空气流速400ml/min,氢气流速40ml/min,尾吹气流速25ml/min。
作为优选,所述步骤(2)的迁移试验中,所用的水基食品模拟物为水、10%乙醇、4%醋酸,其中4%醋酸模拟物需用氢氧化钠调至中性后再进行萃取操作。
与现有技术相比,本发明的优点在于:本发明采用环氧己烷作为内标物质,利用气相色谱内标法对食品接触材料中环氧氯丙烷的迁移量进行测定,该方法可消除操作过程、体积测定等干扰因素,通过加入合适的内标物质,结合操作简单的液液萃取法,建立一种食品接触材料中环氧氯丙烷迁移量的新的检测方法。
本发明中的测定方法过程简单,可操作性强,分析结果具有较好的准确性和可靠性,其中回收率可达89.5%~101.1%,精密度可达2.11~2.83,检出限为0.002mg/l,可为我国食品接触材料中环氧氯丙烷安全评估及检测方法的改进提供基础。
附图说明
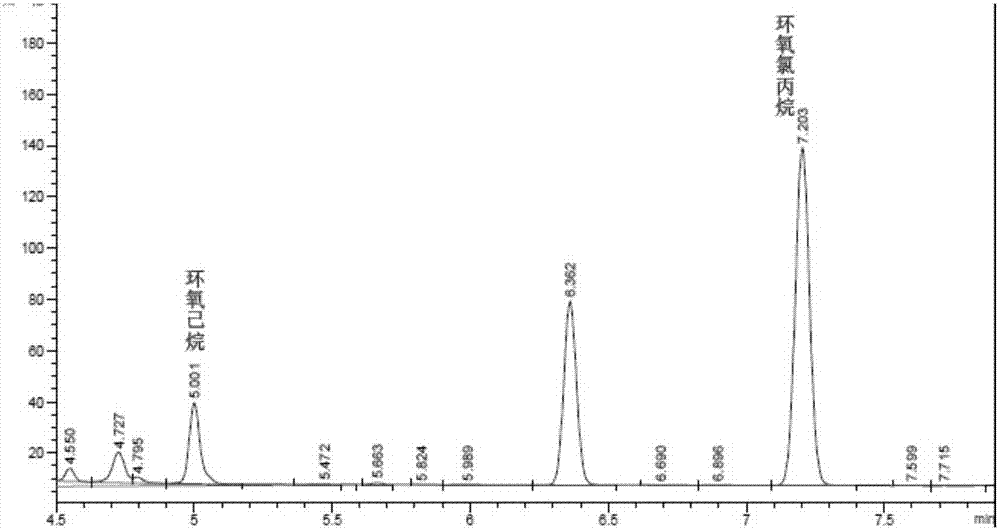
图1为本发明实施例中使用hp-5柱的出峰情况;
图2为本发明实施例中使用hp-innowax的出峰情况。
具体实施方式
以下结合附图实施例对本发明作进一步详细描述。
一、测定方法
1、仪器设备
气相色谱仪:aglient7890(带fid检测器),色谱柱:innowax(30m×0.53mm×1.0μm),hp-5(30m×0.32mm×0.25),高纯氮气、高纯氢气、高纯空气;250ml分液漏斗;振摇仪。
2、试剂
乙酸(分析纯,国药);无水乙醇(分析纯,国药);甲醇(色谱纯,tedia);酚酞指示剂(称取0.5g酚酞溶于50ml95%乙醇,再加纯水50ml);氢氧化钠(分析纯,国药);二氯甲烷(色谱纯,tedia);氯化钠(分析纯,国药);1,2-环氧己烷(纯度≥96%,tgi);环氧氯丙烷(纯度≥95%,德国dr);实验用水为gb/t6682规定的三级水。
3、内标物的选择
环氧氯丙烷的分子量为92,沸点为117.9℃,而环氧己烷也带有环氧基团,且分子量为100,沸点为120℃,不溶于水,溶于醇和二氯化碳,两者的分子结构和物理化学特性较为相近。从仪器响应比较,环氧己烷检测器响应略高于环氧氯丙烷,而极性innowax柱能使目标物质出峰较晚(如图2所示),消除溶剂和模拟物出峰干扰,由图1可见,环氧氯丙烷和环氧己烷的出峰时间分别为5.1min和7.3min,两者接近而又不干扰。因此,本发明选择环氧己烷作为内标物质。
4、色谱条件的选择
4.1色谱柱的选择
环氧氯丙烷检测常用的色谱柱有hp-5、hp-innowax等,通过实验验证了同样程序升温条件下hp-5和hp-innowax的分离效果,得hp-5出峰比较快,出峰时间4min左右(如图1所示)。二氯甲烷使环氧氯丙烷的背景变大,当有乙醇溶液时,则影响环氧氯丙烷的出峰,hp-innowax基本不存在类似影响(如图2所示)。液液萃取前处理都会有少量水存在,hp-innowax柱子能耐受少量水的影响,而且同样浓度条件下,hp-innowax柱子的s/n(信号噪声比)值要比hp-5的s/n值要高。因此本发明选用innowax柱子。
4.2程序升温条件的选择
二氯甲烷、乙醇和环氧氯丙烷的沸点相对较低,因此初始温度越低,与其他物质的分离效果越好。本发明中,试验了若干种程序升温条件,最终确定为:始40℃恒定0.5min,以20℃/min升温速率升温至105℃,恒定4.00min,以40℃/min升温速率升温至200℃,恒定1.00min。
4.3分流比的选择
分流比是模拟物中环氧氯丙烷检测的关键参数,其直接影响了能否对环氧氯丙烷进行准确的测定。在二氯甲烷溶剂中采用不分流进样,以提高检测下限,满足标准限量要求。
由以上试验获得本发明中食品接触材料中环氧氯丙烷迁移量的测定方法,其包括以下步骤:
(1)标准溶液配制:将环氧己烷逐级稀释,用甲醇定容,配制环氧己烷标准中间溶液(500.0mg/l)。将环氧氯丙烷逐级稀释,用甲醇定容,配制环氧氯丙烷标准中间溶液(100.0mg/l)。取适量的环氧氯丙烷标准中间溶液,至10.0ml容量瓶中,并加入环氧己烷中间溶液100μl作内标,用二氯甲烷定容,配制得标准工作液,该标准工作液环氧氯丙烷的浓度范围为0.5、1、2、5、10、20、50mg/l,用于仪器作标准曲线。
(2)样品前处理:准确移取迁移试验得到的模拟液100ml,放入250ml分液漏斗中,加入100μl环氧己烷标准中间溶液(浓度500.0mg/l),加入5g氯化钠,振摇溶解,准确加入5.0ml二氯甲烷,振摇1min。静置分层,滤纸卷成小条,擦干分液漏斗茎管内的水珠,放出下层二氯甲烷萃取液,置于顶空瓶中,盖塞。按照上述步骤,分别准确加入二氯甲烷3ml和2ml,萃取模拟物,合并萃取液于上述同一顶空瓶中,用于气相色谱测定。
(3)气相色谱测定:采用气相色谱法对上述步骤(1)和步骤(2)获得的标准工作液和萃取液分别进行测定,按下列公式(1)计算获得模拟液中的环氧氯丙烷浓度。
式(1)中:
x——模拟液中环氧氯丙烷的含量,单位为mg/l;
ai——萃取液中环氧氯丙烷的峰面积;
ci——标准工作液中环氧氯丙烷的含量,单位为mg/l;
vi——二氯甲烷萃取液体积,单位为ml;
asc——标准工作液中环氧己烷的峰面积;
as——标准工作液中环氧氯丙烷峰面积;
v——模拟液体积,单位为ml;
ass——萃取液中环氧己烷的峰面积,其中vi按等量环氧己烷标准中间溶液定容体积10ml来计算。
上述步骤(3)中气相色谱测定的条件为:色谱柱:hp-innowax,柱温:40℃恒定0.5min,以20℃/min升温速率升温至105℃,恒定4.00min,以40℃/min升温速率升温至200℃,恒定1.00min;气化温度:105℃;载气为n2,其流速及进样方式:恒流,4.0ml/min,不分流;自动进样器进样,进样量1.0μl;fid检测器:温度200℃;空气流速400ml/min,氢气流速40ml/min,尾吹气流速25ml/min。
上述迁移实验根据gb/t5009.156规定,按照待测样品的预期用途和使用条件,用适当的模拟物进行待测样品的迁移试验。本实验以水、10%乙醇、4%醋酸为模拟物按70℃、2小时条件进行迁移实验,分别得到待测模拟物。
二、回收率
1、外标法回收率
(1)标准溶液配制:将环氧己烷逐级稀释,用甲醇定容,配制环氧己烷标准中间溶液(500.0mg/l)。将环氧氯丙烷逐级稀释,用二氯甲烷定容,配制得标准工作液,该标准工作液环氧氯丙烷的浓度范围为0.5、1、2、5、10、20、50mg/l,用于仪器作标准曲线。
(2)样品前处理:准确移取迁移试验得到的水基模拟物100ml,倒入250ml分液漏斗中,加入5g氯化钠,振摇溶解,准确加入5.0ml二氯甲烷,振摇1min。静置分层,滤纸卷成小条,擦干分液漏斗茎管内的水珠,放出下层二氯甲烷萃取液,于顶空瓶中,盖塞。按照上述步骤,分别准确加入二氯甲烷3ml和2ml,萃取模拟物,合并萃取液于同一顶空瓶中(收集前后称重顶空瓶,利用质量差和密度计算二氯甲烷萃取液体积),用气相色谱测定。
(3)气相色谱测定:采用气相色谱法对上述步骤(1)和步骤(2)获得的标准工作液和萃取液分别进行测定,按下列公式(1)计算获得模拟液中的环氧氯丙烷浓度。
外标法按(2)计算:
式(2)中:
x——模拟物中环氧氯丙烷的含量,单位为毫克每升(mg/l);
c——测试液中环氧氯丙烷的浓度,单位为毫克每升(mg/l);
v——模拟物体积,单位为毫升(ml);
vi——二氯甲烷收集体积,单位为毫升(ml)。
在水、10%乙醇和4%乙酸的模拟物溶液中,用环氧氯丙烷标准物质外标法作标准曲线,按式(2)计算,回收率结果如表1所述。
表1外标法回收率(加标量单位mg/l)
2、内标法回收率
在水、10%乙醇和3%乙酸的模拟物溶液中,按上述步骤(1)、(2)、(3)进行操作,在以环氧己烷为内标作环氧氯丙烷标准曲线,按式(1)计算,回收率结果如表2所述。
表2内标法回收率实验(加标量单位mg/l)
对于水基样品采用液液萃取环氧氯丙烷,目前方法成熟,简单可靠,但一些操作过程会对回收率和重现性产生的影响。而采用内标法,则可以避免萃取等过程中的环氧氯丙烷损失造成的定量不准确性,由上述表1和表2,外标法回收率在80.4%~92.3%,而本发明的内标法回收率在89.5%~101.1%,可见内标法的回收率明显优于外标法。
三、精密度试验
在一定加标浓度下分别重复6次,计算rsd得到:在0.5mg/l环氧氯丙烷的3%乙酸模拟液中,外标法rsd(%)为2.97,内标法rsd(%)为2.48;在1.0mg/l环氧氯丙烷的10%乙醇模拟液中,外标法rsd(%)为2.41,内标法rsd(%)为2.11;在2.0mg/l环氧氯丙烷的水模拟液中外标法rsd(%)为5.14,内标法rsd(%)为2.83。在各浓度点内标法精度更高,实验的平行性也更好。
四、线性范围和检出限
本发明对水基食品模拟物的线性范围为0.5~50mg/l,内标法相关系数为0.9996,检出限为0.002mg/l,能够满足欧盟对食品接触材料模拟物中环氧氯丙烷迁移量的限量要求。
- 还没有人留言评论。精彩留言会获得点赞!