一种使用HPLC测定中药一贯煎的指纹图谱方法与流程
本发明涉及中药检测
技术领域:
,尤其涉及一种使用hplc测定中药一贯煎的指纹图谱方法。
背景技术:
:“一贯煎”的处方来源于清代钱敏捷所著《医方絜度》,“一贯煎(柳洲)主肝血衰少,脘痛,胁疼”。“一贯煎”处方组成:北沙参、麦冬、当归各一钱五分,枸杞、生地各三钱,川楝子二钱。“一贯煎”功能主治:具有滋阴疏肝的功效;主治肝肾阴虚,肝气郁滞证,胸脘胁痛,吞酸吐苦,咽干口燥,舌红少津,脉细弱或虚弦,并治疝气瘕聚。现代临床应用主要用于胃溃疡、胃炎、慢性肝炎、肋间神经痛、高血压、神经官能症等的治疗。中药指纹图谱借用dna指纹图谱发展而来。最早发展起来的是中药化学成分色谱指纹图谱,特别是高效液相色谱(hplc)指纹图谱。hplc具有高的分辨率,可将不同的化学成分进行分离而形成高低不同的色谱峰组成一张色谱图,这些色谱峰的高度和峰面积分别代表了各种不同化学成分及其含量。整个色谱图表征了该样品所含化学成分的多少和量的大小。中药指纹图谱在现代及未来的中药质量控制中均占有很重要的地位,符合中医中药整体性和模糊性的特点,其可分析中药中有效成分和无效成分的种类和含量分布的信息,随着越来越多的技术不断被应用于中药指纹图谱的研究,中药指纹图谱必在中药质量控制、中药药效成分研究、中药作用机制研究等多方面发挥更加重要的作用。中药指纹图谱比dna指纹图谱更具优势,不但有特征的体现(各种化学成分的个数和相对位置——保留时间)可作定性鉴别使用,还体现了量的概念,峰的高度和峰面积表征了某个化学成分的含量,而各峰的峰高(或峰面积)的比值体现了各种化学成分间的相对含量。定性和定量的结合赋予中药指纹图谱更大的功效。中药指纹图谱不仅可以进行个体、某物种的“唯一性”的鉴定,还可以将其“量”的特征和其他体系挂钩,例如和药效研究结果挂钩。银杏提取物的指纹图谱体现了其所含33个银杏黄酮(化学成分)和各自含量。德国经30多年的化学成分和药效相关研究,发现约24%银杏黄酮和约6%银杏内酯组成的银杏提取物(有相应的指纹图谱控制其成分和相对含量)具有最好的疗效就是一个典型示例。因此,中药指纹图谱不仅是一种中药质量控制模式和技术,更可以发展成为一种采用各种指纹图谱来进行中药理论(复杂系统)和新药开发的研究体系和研究模式[1]。(参考文献:[1])罗国安,王义明,曹进,多维多息特征谱及其应用[j].中成药,2000,22(6):395)为了控制中药复方一贯煎的质量,如果用一两种当归、生地黄等的活性成分来说明中药一贯煎的内在质量,具有一定的片面性。要控制中药一贯煎的功效,只针对其一两个化学成分进行表征和控制是不够的,必须对它的物质群整体予以控制。在现有的技术中,没有适合快速、简便、精确的分析方法能用于中药一贯煎各种化学成分的个数、相对位置(保留时间)的特征鉴别和含量分布(峰的高度和峰面积表征了某个化学成分的含量,而各峰的峰高(或峰面积)的比值体现了各种化学成分间的相对含量)等相关信息检测。技术实现要素:基于
背景技术:
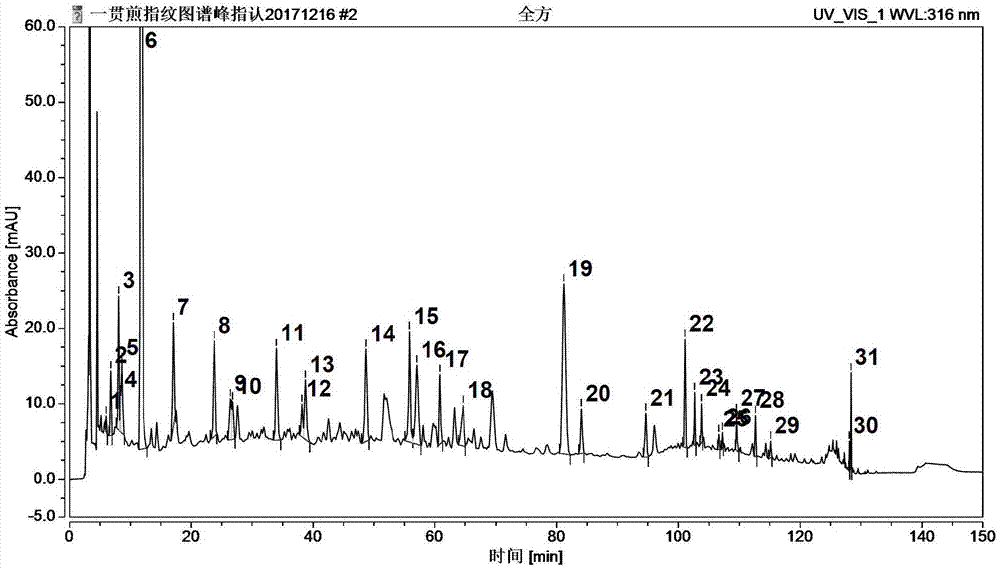
存在的技术问题,本发明提出了一种使用hplc测定中药一贯煎的指纹图谱方法,具有快速、简便、全面、准确可靠的优点。本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以十八烷基硅烷键合硅胶为色谱柱填充剂,以乙腈、甲醇中的一种或两种的混合物为流动相a,以甲酸水溶液、乙酸水溶液、磷酸水溶液中的一种为流动相b,对一贯煎试样溶液进行梯度洗脱,然后采用dad检测器进行检测。优选地,色谱柱内径为4.6mm,色谱柱柱长为250mm。优选地,十八烷基硅烷键合硅胶的粒径为2-10μm。优选地,十八烷基硅烷键合硅胶的粒径为5μm。优选地,选用乙腈为流动相a。优选地,流动相b中,甲酸水溶液的体积浓度为0.01%-1.5%,乙酸水溶液的体积浓度为0.01%-1.5%,磷酸水溶液的体积浓度为0.01%-1.5%。优选地,梯度洗脱过程中,流速为0.5-1.5ml/min,柱温为20-40℃。优选地,梯度洗脱过程中,流速为1.0ml/min,柱温为25℃。优选地,梯度洗脱过程中,流动相a、流动相b的比例变化为:0-10min,a相2%,b相98%;10-90min,a相12%,b相88%;90-120min,a相15%,b相85%;120-125min,a相35%,b相65%;125-135min,a相65%,b相35%;135-140min,a相95%,b相5%;140-150min,a相2%,b相98%。优选地,检测时检测波长为310-320nm。优选地,检测时检测波长为316nm。优选地,一贯煎试样溶液的进样量为8-12μl。优选地,一贯煎试样溶液的进样量为10μl。优选地,一贯煎试样溶液按以下工艺进行制备:在一贯煎干膏粉中加入甲醇,超声10-20min,放冷,滤过,得到一贯煎试样溶液。优选地,在一贯煎干膏粉中加入甲醇,超声10-20min,放冷,滤过,然后将滤液水浴蒸干,加入体积浓度为75-85%的甲醇水溶液溶解,摇匀滤过,得到一贯煎试样溶液。本发明有益效果:与现有技术相比,本发明的有益效果如下:本发明方法通过综合考虑分析色谱柱、流动性、流速、柱温、梯度洗脱程序对分离检测的综合影响,使得检测结果达到了最优化,具有快速、简便、全面、准确可靠的优点,适合检测一贯煎水煎煮液的指纹图谱。本发明中通过优化测定方法,在310-320nm波长处测定,其出峰较多,峰形好,能清楚呈现出当归、生地黄、枸杞子的专属特征峰,易于鉴别,建立的指纹图谱技术含量高,反映的信息较完全,避免了中药一贯煎质量控制的单一性和片面性,能有效反映中药一贯煎的质量;其中,通过对指纹图谱中当归、生地黄、枸杞子专属峰的有无,可有效对中药一贯煎的质量进行评价,保证药材质量的稳定性以及临床用药的有效性和安全性。附图说明图1为本发明实施例1中测得的一贯煎指纹图谱;图2为当归药材指纹图谱;图3为生地黄药材指纹图谱;图4为川楝子药材指纹图谱;图5为枸杞子药材指纹图谱;图6为麦冬药材指纹图谱;图7为北沙参药材指纹图谱。具体实施方式下面,通过具体实施例对本发明的技术方案进行详细说明。实施例1本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为5μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以乙腈为流动相a,以体积浓度为0.01%的甲酸水溶液为流动相b,对10μl一贯煎试样溶液进行梯度洗脱,其中,流速为1.0ml/min,柱温为30℃,然后采用dad检测器进行检测,检测波长为316nm,得到图1。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声15min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为80%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:在相同参数条件下,对一贯煎中单一药材进行测定,得到图2-图7。通过对图2-图7中单一药材的指纹图谱的比照,图1中,当归、生地黄和枸杞子的专属峰号及其保留时间如下:当归专属峰:26号峰(107.240min)、31号峰(128.323min);生地黄专属峰:5号峰(8.617min)、20号峰(84.083min)、21号峰(94.670min)、22号峰(101.110min)、23号峰(102.680min)、25号峰(106.653min)、27号峰(109.530min)、28号峰(112.680min);枸杞子专属峰:12号峰(38.190min)、13号峰(38.737min)、15号峰(55.837min)、17号峰(60.837min)。图1中体现了当归、生地黄、枸杞子的特征峰,而北沙参、麦冬、川楝子在该色谱条件下基本检不出特征峰;采用本发明实施例1中检测方法,结合图1发现,在316nm波长处测定中药一贯煎,其出峰较多,峰形好,能清楚呈现出当归、生地黄、枸杞子的专属特征峰,易于鉴别,建立的指纹图谱技术含量高,反映的信息较完全,避免了中药一贯煎质量控制的单一性和片面性,能有效反映中药一贯煎的质量,从而能保证药材质量的稳定性以及临床用药的有效性和安全性。实施例2本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为5μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以乙腈为流动相a,以体积浓度为0.1%的乙酸水溶液为流动相b,对一贯煎试样溶液进行梯度洗脱,其中,流速为1.0ml/min,柱温为30℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置具塞锥形瓶中,加入甲醇40ml,超声15min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为80%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表2所示:时间(min)流动相a%流动相b%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例3本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为5μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以乙腈为流动相a,以体积浓度为0.1%的磷酸水溶液为流动相b,对一贯煎试样溶液进行梯度洗脱,其中,流速为1.0ml/min,柱温为30℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置具塞锥形瓶中,加入甲醇40ml,超声15min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为80%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表2所示:时间(min)流动相a%流动相b%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例4本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为5μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm,型号为shim-packvp-ods;以乙腈为流动相a,以体积浓度为0.1%的磷酸水溶液为流动相b,对一贯煎试样溶液进行梯度洗脱,其中,流速为1.0ml/min,柱温为30℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置具塞锥形瓶中,加入甲醇20ml,超声15min,放冷,滤过,得到一贯煎试样溶液。梯度洗脱过程中,流动相a、流动相b的比例变化如表2所示:时间(min)流动相a%流动相b%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例5本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为5μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm,型号为agilent5hc-c18(2);以乙腈为流动相a,以体积浓度为0.1%的磷酸水溶液为流动相b,对一贯煎试样溶液进行梯度洗脱,其中,流速为1.0ml/min,柱温为30℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置具塞锥形瓶中,加入甲醇20ml,超声15min,放冷,滤过,得到一贯煎试样溶液。梯度洗脱过程中,流动相a、流动相b的比例变化如表2所示:时间(min)流动相a%流动相b%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例6本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为2μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以甲醇为流动相a,以体积浓度为1.5%的甲酸水溶液为流动相b,对8μl一贯煎试样溶液进行梯度洗脱,其中,流速为1.5ml/min,柱温为40℃,然后采用dad检测器进行检测,检测波长为310nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声10min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为85%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:时间(min)流动相a(乙腈)%流动相b(0.1%磷酸)%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例7本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为10μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以甲醇-乙腈混合液为流动相a,以体积浓度为0.01%的乙酸水溶液为流动相b,对12μl一贯煎试样溶液进行梯度洗脱,其中,流速为0.5ml/min,柱温为20℃,然后采用dad检测器进行检测,检测波长为320nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声20min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为75%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:时间(min)流动相a(乙腈)%流动相b(0.1%磷酸)%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例8本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为6μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以甲醇-乙腈混合液为流动相a,以体积浓度为1.5%的乙酸水溶液为流动相b,对10μl一贯煎试样溶液进行梯度洗脱,其中,流速为1.2ml/min,柱温为25℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声15min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为75%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:时间(min)流动相a(乙腈)%流动相b(0.1%磷酸)%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例9本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为10μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以甲醇为流动相a,以体积浓度为0.01%的磷酸水溶液为流动相b,对10μl一贯煎试样溶液进行梯度洗脱,其中,流速为0.8ml/min,柱温为25℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声20min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为75%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:时间(min)流动相a(乙腈)%流动相b(0.1%磷酸)%0-1029810-90109090-1201585120-1253565125-1357525135-140955140-150298实施例10本发明提出的一种使用hplc测定中药一贯煎的指纹图谱方法,包括以下步骤:以粒径为10μm的十八烷基硅烷键合硅胶为色谱柱填充剂,色谱柱内径为4.6mm,色谱柱柱长为250mm;以甲醇为流动相a,以体积浓度为1.5%的磷酸水溶液为流动相b,对10μl一贯煎试样溶液进行梯度洗脱,其中,流速为0.8ml/min,柱温为25℃,然后采用dad检测器进行检测,检测波长为316nm。其中,一贯煎试样溶液按以下工艺进行制备:称取一贯煎干膏粉2.0g置于具塞锥形瓶中,加入甲醇40ml,超声15min,放冷,滤过,滤液置干燥洁净的烧杯中,水浴蒸干,以体积浓度为80%的甲醇水溶液溶解并定容至5ml容量瓶中,摇匀滤过,得到一贯煎试样溶液;梯度洗脱过程中,流动相a、流动相b的比例变化如表1所示:以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1