测定植物样品中羟基多溴联苯醚的方法与流程
本发明涉及检测技术领域,尤其涉及测定植物样品中羟基多溴联苯醚的方法。
背景技术:
多溴联苯醚(pbdes)一种重要的新型溴代阻燃剂,因其阻燃效率高和热稳定性好、对材料性能影响小等特性,广泛应用于塑料、电器、家具、纺织品和化工等领域;同时pbdes是一类环境中广泛存在的全球性有机污染物,一方面是因为pbdes在室温下具有蒸汽压低和亲脂性强的特点,可以散逸到空气中,随大气长距离传输,另一方面是因为其化学性质稳定,可能随着食物链生物富集和放大,具有生物可累积性,目前,已经在各种环境介质(大气、沉积物、土壤、生物体等)以及人体血清和母乳中检测出pbdes。
环境中的pbdes可以发生降解代谢,如脱溴还原代谢和氧化代谢(羟基化和甲氧基化代谢),其中羟基多溴联苯醚(oh-pbdes)除与pbdes具有类似的毒性作用外,还具有影响雌二醇合成,干扰氧化磷酸化(oxphos)作用等毒理特征,最主要毒性效应是内分泌干扰,如干扰甲状腺激素体内平衡、干扰甾类的产生以及毒害神经元效应,oh-pbdes的主要来源一方面来自于自然环境,另一方面来自于环境中pbdes的转化,由于oh-pbdes具有比pbdes更大的毒性效应,因此也越来越引起关注。
目前对生物样品中oh-pbdes的检测方法主要采用气相色谱–质谱联用法分析其衍生化产物的甲氧基多溴联苯醚(meo-pbdes),采用gc–ms分析虽然灵敏性高,但必须的衍生化步骤非常耗时,样品的前处理很繁琐,同时衍生化试剂通常都比较危险,且不完全衍生化也会导致定量结果不准确,近年来,由于液相色谱–质谱联用法(lc-ms)不需要衍生化处理,以及具有高灵敏度、优越的分离能力和简单快速的优点,在环境分析领域的应用越来越广,逐渐成为一种重要的分析手段。但是已有的关于oh-pbdes的lc-ms分析方法主要是针对动物组织和血液样品,而对植物样品的提取和测定还未见报道,另外对于植物样品中oh-pbdes的检测,由于植物样品中往往会存在大量的叶绿素和脂类等杂质,会影响oh-pbdes在质谱检测中的结果,因此在前处理部分能够有效地将叶绿素及脂类等杂质去除是测定植物中oh-pbdes含量的前提,现有的去除叶绿素的方法大多采用活性炭吸附的方法,但是活性炭吸附时选择性不高,容易将其它有用的物质也吸附,造成分析结果不准确。
技术实现要素:
有鉴于此,本发明的目的是提供测定植物样品中羟基多溴联苯醚的方法,以超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪联合分析的方法,简便快速、灵敏度高,且在处理过程中采用磁性分子印迹材料作为选择性吸附剂,选择性较高,吸附效果较好。
本发明通过以下技术手段解决上述技术问题:
测定植物样品中羟基多溴联苯醚的方法,包括以下步骤:
提取:将经过冷冻干燥后的植物样品剪碎,加入37wt%的hcl溶液和异丙醇混匀,再加入正己烷-mtbe混合溶液a,超声震荡24h,离心,移出一次提取有机相,沉淀重复前述所有操作,合并两次一次提取有机相得到粗提取液;
二次提取:将粗提取液氮吹吹干后,溶解于正己烷中,加入naoh溶液萃取,分离得到二次提取有机相和水相,水相中加入正己烷再次萃取水相,再加入hcl溶液,并用正己烷-mtbe混合溶液b提取三次后,合并所有二次提取有机相,得到提取液;
叶绿素去除:向得到的提取液中加入选择吸附剂超声震荡30min后,静置24h,过滤,得到粗提纯液;
提纯:将粗提纯液过弗罗里硅土柱,用二氯甲烷-正己烷-甲醇混合溶液预淋洗,再用二氯甲烷-正己烷-甲醇混合溶液洗脱后,得到的洗脱液即为提纯液;
分析:将提纯液经氮吹吹干后定容,过0.22μm滤膜后进行超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪分析。
通过提取-二次提取-叶绿素去除-提纯等前处理步骤,将植物样品中的羟基多溴联苯醚提取出来,通过物理吸附技术将植物样品中大量存在叶绿素去除,同时不会对目标物质进行吸附,在一定程度上减少对其检测结果的影响,且前处理过程无需衍生化,能获得较低的检出限和回收率,超高效液相色谱–质谱联用技术简便快速、能够很好的分离oh-pbdes同系物和同分异构体,并可以准确对其定性和定量分析,同时具有较高的灵敏度。
进一步,所述叶绿素去除步骤中,所述选择吸附剂是以磁性纳米粒子为核心,β-环糊精为单体,血红素为模板分子的磁性分子印迹材料。
以磁性分子印迹材料作为选择吸附剂,以血红素为模板,对叶绿素有较高的选择性,不会将提取液中其它需要的目的物质吸附,减少前处理过程中目标物质的损失。
进一步,所述磁性纳米粒子以多孔磁性四氧化三铁为核心,外包覆了二氧化硅/聚氯甲基苯乙烯复合保护层的球形粒子,所述保护层表面形成若干针状凸起。
该结构的选择性吸附剂,具有较大的比表面积,因此结合的位点更多,吸附的饱和度更高,效果更好,另外在磁性多孔磁性四氧化三铁的核心外包裹了二氧化硅/聚氯甲基苯乙烯的复合层,使得选择吸附剂的强度更高,耐酸碱性更好,稳定性更好。
进一步,所述提取步骤中,hcl溶液和异丙醇的体积比为1:5,所述正己烷-mtbe混合溶液a中正己烷与mtbe的体积比为1:1。
进一步,所述二次提取步骤中,naoh溶液为0.5mol/lnaoh溶于50%乙醇中得到,所述正己烷-mtbe混合溶液b中正己烷与mtbe的体积比为9:1。
进一步,所述提纯步骤中使用的弗罗里硅土柱为10mmi.d.,从上到下为3.0cm无水硫酸钠和5.0cm弗罗里硅土。
进一步,所述超高效液相色谱采用c18柱,以乙腈和水作为流动相,设置柱温40℃,样品室温度10℃,进样量10μl,采用梯度洗脱程序。
进一步,所述电喷雾离子源设置毛细管电压2.5kv,离子源温度150℃,去溶剂温度500℃,去溶剂气流量700l/h,锥孔气流量50l/h,碰撞气体选用氦气。
进一步,所述三重四级杆质谱仪分析中设置锥孔电压22-40v,碰撞气电压20-25v。
本发明的有益效果:
(1)本发明通过提取-二次提取-叶绿素去除-提纯等前处理步骤,将植物样品中的羟基多溴联苯醚提取出来,过程无需衍生化,能获得较低的检出限和回收率,通过物理吸附技术将植物样品中大量存在叶绿素去除,同时不会对目标物质进行吸附,在一定程度上减少处理过程对检测结果的影响,提高检测结果的准确度。
(2)本发明的分析方法采用超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪的分析方法,简便快速、能够很好的分离oh-pbdes同系物和同分异构体,并可以准确对其定性和定量分析,同时具有较高的灵敏度。
(3)以磁性分子印迹材料作为选择吸附剂,选择性较好,以多孔磁性四氧化三铁为核心,外包覆二氧化硅/聚氯甲基苯乙烯复合层,且在保护层表面形成若干针状凸起,该结构具有较大的比表面积,因此该选择吸附剂的吸附饱和度更高,且强度更高,耐酸碱性更好,稳定性更好。
附图说明
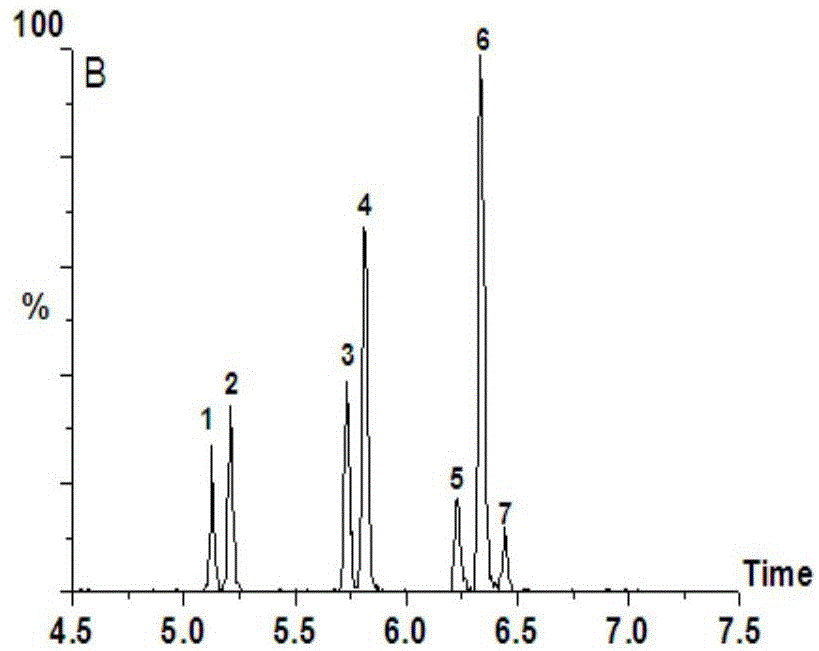
图1是本发明实施例一中超高效液相色谱分析所得超高效液相色谱图;
其中,1.2'-oh-bde3、2.3'-oh-bde7、3.4'-oh-bde17、4.3'-oh-bde28、5.3-oh-bde47、6.5-oh-bde47、7.6-oh-bde47。
具体实施方式
以下将结合具体实施例对本发明进行详细说明:
本发明的测定植物样品中羟基多溴联苯醚的方法中所使用的选择吸附剂的制备方法如下:
多孔磁性四氧化三铁的制备:在氮气保护下,取20g1.0mmol/g的feso4·7h2o和40g2.0mmol/g的feso4·6h2o加入1000ml乙二醇中,搅拌至固体溶解后,加入100g十二烷胺(表面活性剂),在300rpm、25℃的条件下搅拌10min,加入20g醋酸铵,继续搅拌30min,然后继续搅拌加入15g柠檬酸钠,于500rpm的条件下搅拌至悬浮液变成均匀溶液,将其置于不锈钢反应釜中,将反应釜放置在烘箱中加热至200℃,并保温10h,之后降温到室温,然后以10000r/min的速度离心5min,以乙醇和去离子水洗涤,在真空度为-0.06mpa,温度为60℃的条件下真空干燥,得到多孔磁性四氧化三铁;
包覆:取60g多孔磁性四氧化三铁置于1000ml甲醇溶液中,再加入180g针状的纳米碳酸钡粉末,搅拌12h后,继续于频率30khz、功率100w条件下超声波填充15min,使针状的纳米碳酸钡插接到多孔磁性纳米四氧化三铁粒子的孔隙中,填充完成后,然后在外加磁场作用下分离沉淀,并对分离出来的沉淀用乙醇进行洗涤后,将其分散于2000ml乙醇溶液中,搅拌,然后加入500ml正硅酸乙酯,继续搅拌1h后,缓慢滴加1000ml25%的氨水溶液,继续搅拌反应24h后,在外加磁场的作用下再次分离沉淀,对分离得到的沉淀进行洗涤后,将其分散到1000ml甲苯溶液中,再加入300ml对氯甲基苯乙烯和20g偶氮二异丁基,水浴加热至70℃,于17rpm搅拌反应13h,反应完成后,冷却到室温,加入500ml甲醇溶液,在冰浴下搅拌1-2h,冷却完成后在外加磁场的作用下再次分离沉淀,对分离得到的沉淀以甲醇和去离子水多次洗涤后,在真空度为-0.06mpa,温度为70℃的条件下真空干燥,得到磁性纳米粒子。
修饰:取60g磁性纳米粒子加入1500ml0.1mol/l的hcl溶液中,于频率25khz、功率100w条件下超声振荡分散10min,然后经外磁场分离、去离子水洗涤后分散于乙醇-去离子水-浓氨水的混合溶液中,然后加入240gβ-环糊精,以100rpm的搅拌速度,在室温下反应6h,离心、洗涤,在真空度为-0.06mpa,温度为65℃的条件下真空干燥之后得到以β-环糊精修饰的磁性纳米粒子;
分子印迹处理:取60g血红素溶解于1000ml稀氨水溶液中,再加入5g3-(2-氨基乙基氨基)丙基三甲氧基硅烷和5g偶氮二异丁腈,搅拌30min后,得到混合溶液,再将以β-环糊精修饰的磁性纳米粒子分散于1000ml甲醇溶液中,随后加入到所述混合溶液中,继续搅拌,于70℃下反应18h后,分离;
洗脱:将经分子印迹处理步骤得到的固体用去离子水和甲醇分别洗涤,直至洗脱液的ph=6-7,真空干燥后得到选择吸附剂。
本发明的测定植物样品中羟基多溴联苯醚的方法如下:
实施例一
提取:将经过冷冻干燥后的植物样品剪碎,准确称取3.0g(干重),加入2ml、37wt%的hcl溶液和10ml异丙醇混匀,再加入3ml正己烷-mtbe混合溶液a(正己烷-mtbe混合溶液a中正己烷与mtbe的体积比为1:1),超声震荡24h,于10000rpm的速度下离心,移出一次提取有机相,沉淀重复前述所有操作,合并两次一次提取有机相得到粗提取液;
二次提取:将粗提取液中的有机相氮吹吹干后,溶解于2ml正己烷中,加入1mlnaoh溶液萃取(其中naoh溶液由0.5mol/lnaoh溶于50%乙醇中得到),分离得到二次提取有机相和水相,水相中加入2ml正己烷再次萃取水相,再加入300μl、2mhcl溶液,并用1ml正己烷-mtbe混合溶液b提取三次后(其中正己烷-mtbe混合溶液b中正己烷与mtbe的体积比为9:1),合并所有二次提取有机相,得到提取液;
叶绿素去除:向得到的提取液中加入0.2g选择吸附剂超声震荡30min后,静置24h,过滤,得到粗提纯液;
提纯:将粗提纯液过弗罗里硅土柱,弗罗里硅土柱采用10mmi.d.,从上到下为3.0cm无水硫酸钠和5.0cm弗罗里硅土的柱子,用10ml二氯甲烷-正己烷-甲醇混合溶液预淋洗,再用25ml二氯甲烷-正己烷-甲醇混合溶液洗脱后,得到的洗脱液即为提纯液,其中二氯甲烷-正己烷-甲醇混合溶液中二氯甲烷:正己烷:甲醇为50:45:5;
分析:将提纯液经氮吹吹干后用乙腈水溶液(乙腈:水=50:50)定容至300μl,过0.22μm滤膜后进行超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪分析。
实施例二
提取:将经过冷冻干燥后的植物样品剪碎,准确称取4.0g(干重),加入3ml、37wt%的hcl溶液和15ml异丙醇混匀,再加入4.5ml正己烷-mtbe混合溶液a(正己烷-mtbe混合溶液a中正己烷与mtbe的体积比为1:1),超声震荡24h,于10000rpm的速度下离心,移出一次提取有机相,沉淀重复前述所有操作,合并两次一次提取有机相得到粗提取液;
二次提取:将粗提取液中的有机相氮吹吹干后,溶解于3ml正己烷中,加入1.5mlnaoh溶液萃取(其中naoh溶液由0.5mol/lnaoh溶于50%乙醇中得到),分离得到二次提取有机相和水相,水相中加入3ml正己烷再次萃取水相,再加入300μl、2mhcl溶液,并用2ml正己烷-mtbe混合溶液b提取三次后(其中正己烷-mtbe混合溶液b中正己烷与mtbe的体积比为9:1),合并所有二次提取有机相,得到提取液;
叶绿素去除:向得到的提取液中加入0.2g选择吸附剂超声震荡30min后,静置24h,过滤,得到粗提纯液;
提纯:将粗提纯液过弗罗里硅土柱,弗罗里硅土柱采用10mmi.d.,从上到下为3.0cm无水硫酸钠和5.0cm弗罗里硅土的柱子,用15ml二氯甲烷-正己烷-甲醇混合溶液预淋洗,再用40ml二氯甲烷-正己烷-甲醇混合溶液洗脱后,得到的洗脱液即为提纯液,其中二氯甲烷-正己烷-甲醇混合溶液中二氯甲烷:正己烷:甲醇为50:45:5;
分析:将提纯液经氮吹吹干后用乙腈水溶液(乙腈:水=50:50)定容至300μl,过0.22μm滤膜后进行超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪分析。
实施例三
提取:将经过冷冻干燥后的植物样品剪碎,准确称取5.0g(干重),加入4ml、37wt%的hcl溶液和20ml异丙醇混匀,再加入5ml正己烷-mtbe混合溶液a(正己烷-mtbe混合溶液a中正己烷与mtbe的体积比为1:1),超声震荡24h,于10000rpm的速度下离心,移出一次提取有机相,沉淀重复前述所有操作,合并两次一次提取有机相得到粗提取液;
二次提取:将粗提取液中的有机相氮吹吹干后,溶解于4ml正己烷中,加入3mlnaoh溶液萃取(其中naoh溶液由0.5mol/lnaoh溶于50%乙醇中得到),分离得到二次提取有机相和水相,水相中加入4ml正己烷再次萃取水相,再加入300μl、2mhcl溶液,并用3ml正己烷-mtbe混合溶液b提取三次后(其中正己烷-mtbe混合溶液b中正己烷与mtbe的体积比为9:1),合并所有二次提取有机相,得到提取液;
叶绿素去除:向得到的提取液中加入0.4g选择吸附剂超声震荡30min后,静置24h,过滤,得到粗提纯液;
提纯:将粗提纯液过弗罗里硅土柱,弗罗里硅土柱采用10mmi.d.,从上到下为3.0cm无水硫酸钠和5.0cm弗罗里硅土的柱子,用20ml二氯甲烷-正己烷-甲醇混合溶液预淋洗,再用50ml二氯甲烷-正己烷-甲醇混合溶液洗脱后,得到的洗脱液即为提纯液,其中二氯甲烷-正己烷-甲醇混合溶液中二氯甲烷:正己烷:甲醇为50:45:5;
分析:将提纯液经氮吹吹干后用乙腈水溶液(乙腈:水=50:50)定容至300μl,过0.22μm滤膜后进行超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪分析。
在以上所有实施例中,进行超高效液相色谱–电喷雾离子源–串联三重四级杆质谱仪分析时,超高效液相色谱采用c18柱,以乙腈和水作为流动相,设置柱温40℃,样品室温度10℃,进样量10μl,采用梯度洗脱程序,梯度洗脱程序见表1:
表1梯度洗脱条件
采用线性梯度洗脱的方式能获得较好的色谱峰形和分离效果,其中对实施例一进行超高效液相色谱检测所得结果如图1所示。
电喷雾离子源设置毛细管电压2.5kv,离子源温度150℃,去溶剂温度500℃,去溶剂气流量700l/h,锥孔气流量50l/h,碰撞气体选用氦气,采集模式为多反应检测,使用masslynxtm4.1软件获取数据,其它质谱分析参数见表2:
表2质谱检测条件
在三重四级杆质谱仪分析中设置锥孔电压22-40v,碰撞气电压20-25v。
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。本发明未详细描述的技术、形状、构造部分均为公知技术。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>GeO<sub>5</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiOBr/BiOI复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/MgFe<sub>2</sub>O<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>/BiVO<sub>4</sub>复合环境催化材料的制备方法