一种非模式生物转录组基因序列结构分析的方法与流程
本发明涉及基因分析生物信息学领域,具体是一种非模式生物转录组基因序列结构分析的方法。
背景技术:
基因是细胞内具有生物学功能的一段核酸序列,其翻译的蛋白质直接参与了细胞的各种生理生化反应,是细胞赖以生存的基础。近些年生命科学的研究的其中一个最主要的方向就是基因功能探索,所以很多生物基础的研究的起点就是确定该物种的基因数目和类型。传统的基因同源克隆为生物基因序列的获得和研究提供了可靠的研究方法,但是基因克隆只能对单个基因序列逐一进行,很难对细胞内所有的基因序列进行高通量的测序。最新的高通量测序技术在转录组学上的应用,转录组测序为细胞内所有的基因序列的大规模测定提供了新的方法,但是这种大规模测序又为海量基因序列的准确分析提出了新的挑战。如何确定转录组测序拼接得到的大量基因,特别是蛋白编码基因的序列结构是后续基因功能分析的基础。
基因作为生物遗传的基本功能单元,其在各种生物体细胞内广泛存在,且不同物种的基因由于物种进化的关系,存在着广泛的相似和同源性。基于此,为了确定转录组拼接得到的基因种类,最常用的基因注释方法就是将序列比对到已知的物种的核酸和蛋白序列,根据同源比对的方法,通过已知的同源序列的功能推测未知的序列的种类。但是这种基因注释只能确定序列的功能,还不能对序列的结构,特别是5’和3’非编码区(Un-Translated Region,5’-,3’-UTR)和编码区(CoDing Sequences,CDS)进行准确地确定;而UTR和CDS的确定对于基因功能的研究非常重要,因为很多基因的调控就是通过UTR区域实现的。比如想要了解某个基因受到miRNA的调控,最直接的一个方法就是将基因的UTR序列与该物种的miRNA的种子区域进行比对。另外的一个应用就是对突变位点的生物功能的确定,就序列明确基因编码蛋白的方式、起始和终止坐标,才能判断该突变是不是在编码区,会不会导致蛋白质的变化等等。
对于大量的非模式生物而言,没有任何关于该物种的公共的相关基因序列信息,其转录组拼接的基因的结构分析更加困难。针对这种情况,目前基因结构分析的比较常用的方法之一就是最长编码算法,比如NCBI的ORFFinder程序。该算法自动寻找使用者提供的核酸序列的6中编码方式中,寻找起始密码子和终止密码子,找到最长的可编码基因为其最可能编码的序列。这个方法最大的特点是能够找到的最长的翻译的蛋白,运行迅速,但是该方法不能对翻译的蛋白的功能做任何保证,并且最长的编码也不一定就是基因编码的蛋白序列,所以该方法往往具有较高的假阳性。为了克服最长编码算法的假阳性,生物信息学家们提出使用马尔科夫链的方法对基因的编码方式进行确认。其主要的思路是利用编码最长的蛋白的前100-500条基因序列作为训练集,训练该物种的核酸编码蛋白的马尔科夫链模型,然后利用该模型对该物种的所有核酸序列的6种编码方式计算其概率,概率最高的为最可能的蛋白编码方式,在确定该编码方式下的起始密码子和终止密码子,从而对大量的基因序列进行结构分析。这种方法考虑到了物种特异性,使用物种的蛋白编码的基因序列构建模型,在一定程度上提高了预测的准确度,但是这种方法仍然不能保证预测的翻译的蛋白的功能。
技术实现要素:
本发明的目的在于提供一种能大幅提高基因编码方式的预测准确度,并能保证大部分基因序列翻译的蛋白质功能的非模式生物转录组基因序列结构分析的方法,以解决上述背景技术中提出的现有的大规模基因序列结构分析方法假阳性高,且无法保证翻译的蛋白序列功能的问题。
为实现上述目的,本发明提供如下技术方案:
一种非模式生物转录组基因序列结构分析的方法,包括以下步骤:
(1)通过序列比对,得到转录组基因序列在公共蛋白数据库的最优比对结果;
(2)根据比对结果,确定有比对结果的基因序列的蛋白编码模式,确定翻译终止位置;
(3)利用公共的蛋白序列,通过马尔科夫链训练获得编码起始的序列特征,确定基因序列的编码起始位置;
(4)使用已知的编码蛋白的核酸序列,利用支持向量机SVM训练编码蛋白的基因模型,对于没有比对上任何已知蛋白序列的基因利用上述模型进行分类;
(5)使用转录组序列中确定编码方式的核酸序列,使用马尔科夫链训练编码蛋白的核酸序列模型;
(6)对于通过SVM分类为蛋白编码的核酸序列,通过上述马尔科夫链模型,确定剩余蛋白编码序列的编码方式。
作为本发明进一步的方案:步骤(1)中以公共蛋白质数据库为参考,使用blastx程序将转录组拼接得到的核酸序列比对到蛋白数据库中;对于每个核酸序列,只保留其最佳的比对结果:E值最小的比对。
作为本发明再进一步的方案:步骤(2)中将蛋白比对的结果转换为核酸的比对,确定核酸翻译蛋白序列的编码方式,并按照这个编码方式向后继续读取核酸三联体密码子,一直读到终止密码子结束;如果没有读到终止密码,则表示该cDNA序列不完整,核酸序列全部翻译直到序列结束;如果读到终止密码,则后续的序列是该基因的3’-UTR。
作为本发明再进一步的方案:步骤(3)中利用所述述公共蛋白序列库,分别利用马尔科夫链训练起始氨基酸甲硫氨酸Met后续的氨基酸序列和非起始甲硫氨酸后续的氨基酸序列模型;利用步骤(2)中获得的核酸编码方式,向前提取三联体密码子,如果碰到Met,则利用Met后的核酸序列判断该Met为起始密码子的可能性。
作为本发明再进一步的方案:步骤(4)中利用步骤(3)中获得的编码蛋白的序列,利用多种基因序列和表达量特征构建蛋白编码基因的SVM模型,对于步骤(1)中未比对上任何蛋白的核酸序列,使用SVM模型模型进行分类,显著判定为蛋白编码的序列进行步骤(5)的分析。
作为本发明再进一步的方案:步骤(5)中利用步骤(3)中获得的核酸编码的马尔科夫模型,对步骤(4)中判定为蛋白编码的核酸序列预测最有可能的蛋白编码开放阅读框模式,并分别向前向后寻找起始和终止密码子;对于Met是否为起始密码子的确定,使用步骤(3)中的方法进行判断。
与现有技术相比,本发明的有益效果是:本发明提供了一种生物转录组基因序列结构分析的方法,能够对任何非模式生物的转录组测序获得的大量的基因序列进行高通量结构分析。相对于现有的分析技术,该方法具有以下几个优点:
(1) 不受研究物种的限制,只要相关其他物种的蛋白序列支持,就可以对任何物种的转录组基因序列进行结构分析;
(2)该分析过程自动完成了转录组序列的注释,可以与现有的转录组注释流程整合,丰富现有的转录组拼接得到的核酸序列功能注释的内涵;
(3)由于本发明的方法基于的是核酸序列的公共蛋白数据库比对,因而获得的蛋白序列大部分都是具有明确生物学功能的氨基酸序列;
(4)本发明的方法是利用基于比对的高度可靠的蛋白编码核酸序列构建了马尔科夫模型和支持向量机模型,相对于其他的方法其构建模型的基础序列数据的可信度更高;
(5) 本发明的方法对于没有任何比对的核酸序列也应用了支持向量机对其蛋白编码性进行预测,为物种特异基因的后续研究提供了重要的序列信息。
附图说明
图1为非模式生物转录组基因序列结构分析的方法的分析流程示意图。
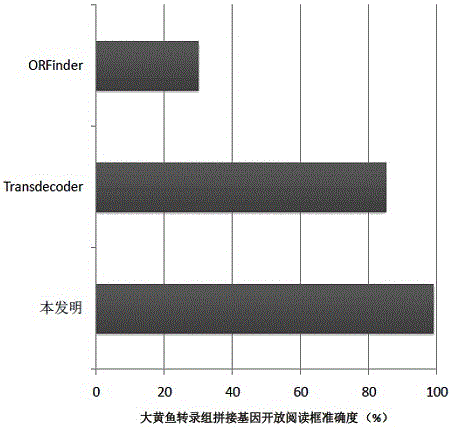
图2为非模式生物转录组基因序列结构分析的方法中中大黄鱼转录组拼接的基因序列结构分析结果比较图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
请参阅图1~2,本实施例利用该发明提供的一种非模式生物转录组基因序列结构分析的方法,对大黄鱼的转录组拼接产物进行基因序列结构分析。
大黄鱼(Larimichthys crocea),俗称黄鱼、黄花鱼,隶属硬骨鱼纲鲈形目石首鱼科黄鱼属,是我国近海重要经济鱼类,有“海水国鱼”之称。目前,大黄鱼是我国育苗和养殖量最多的海水鱼类之一,年产量已经超过12万吨,每年的直接经济产值数十亿元。对大黄鱼的基因序列进行全面分析,是大黄鱼遗传研究重要的遗传资源,是探讨生长速度,肌肉品质和抗病性等重要经济性状性状遗传基因的基础,也为后续进行分子辅助育种和全基因组关联分析的提供了重要的依据。本实例利用本发明提供的非模式生物转录组基因序列结构分析的方法,对大黄鱼转录组测序拼接得到的37511条基因序列进行分析,以解释本发明的具体分析步骤和方法。为了检验为了说明本方法的准确度,本实例使用人的公共参考蛋白序列进行分析,包括以下步骤:
(1) 通过公共蛋白数据库序列的局部比对,得到转录组基因序列的最优比对结果。在ensembl公共数据库下载斑马鱼的全长参考蛋白序列(版本号GRCz10)。使用blast+软件包(版本号2.4.0)的makeblastdb使用人的蛋白数据构建搜索数据库库,并使用blastx程序将大黄鱼的37511条基因序列比对到人的蛋白数据库上。比对的主要参数如下:-evalue 1e-5 –num_threads 32。按照E值对每一条大黄鱼基因序列选择最佳的目标序列,得到32135条序列的最佳比对;
(2)根据序列比对确定基因序列的蛋白编码模式,确定翻译终止位置。使用步骤(1)中的比对结果,确定有比对结果的大黄鱼基因序列翻译蛋白的开放阅读框的编码方式,并在大黄鱼基因组序列上向后延伸三联体密码。32135条有比对的大黄鱼序列中,有21591条成功找到终止密码子序列,确定翻译终止位置,其后的序列为这些基因的3’-UTR;剩余的10544条序列没有找到终止密码,则一直翻译到序列末;
(3)利用人的蛋白序列数据,通过马尔科夫链训练获得编码起始的序列特征,确定基因序列的编码起始位置。在人的蛋白序列数据库中,分别确定Met为起始氨基酸和非起始氨基酸的后续序列,并分别利用马尔科夫链构建起始Met和非起始Met的模型。在步骤(2)中有比对的序列中,向前延伸三联体密码子,寻找起始密码子(ATG)。如果发现密码子翻译Met,则使用上述马尔科夫模型判断该Met是否为起始氨基酸。如果按照Met为起始氨基酸的模型计算的概率较高,则认为该ATG为翻译起始位点,否则则继续向前延伸蛋白序列,直到找到翻译起始位点或者序列结束。按照这个方法,32135条有比对的大黄鱼序列中,19856条序列成功找到翻译起始位点;
(4)使用已知的编码蛋白的核酸序列,利用支持向量机(SVM)训练编码蛋白的基因模型,对于没有比对上任何已知蛋白序列的基因利用上述模型进行分类。为了进一步对为比对上任何人类蛋白的基因进行分析,利用步骤(2)和(3)中找到的32135条编码蛋白的大黄鱼序列构建SVM模型。使用的基因序列属性包括:序列长度,GC含量,CPAT软件预测分数,基因表达量(FPKM)。使用22135条序列进行模型训练,10000条序列进行模型验证,发现该SVM模型的准确度高达98.3%。对步骤1中未比对上任何人蛋白序列的5376条大黄鱼基因序列,使用该SVM模型进行预测,发现其中3290条序列为蛋白编码序列。
(5)对于步骤(4)中通过SVM分类为蛋白编码的3290条核酸序列,利用步骤(2)的方法和步骤(3)中构建的马尔科夫链模型,确定这些蛋白编码序列的编码方式。
(6)对于上述5步中获得的大黄鱼转录组序列可翻译蛋白序列的编码方式,利用大黄鱼全基因组注释信息提供的蛋白序列进行一一比对,验证蛋白编码方式检测的准确性。并分别使用ORFinder和transdecoder进行分析,比较蛋白编码方式判断的准确度。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
- 还没有人留言评论。精彩留言会获得点赞!