一种构建miRNA调控网络的方法与流程
本发明属于数据处理
技术领域:
,尤其涉及一种构建mirna调控网络的方法。
背景技术:
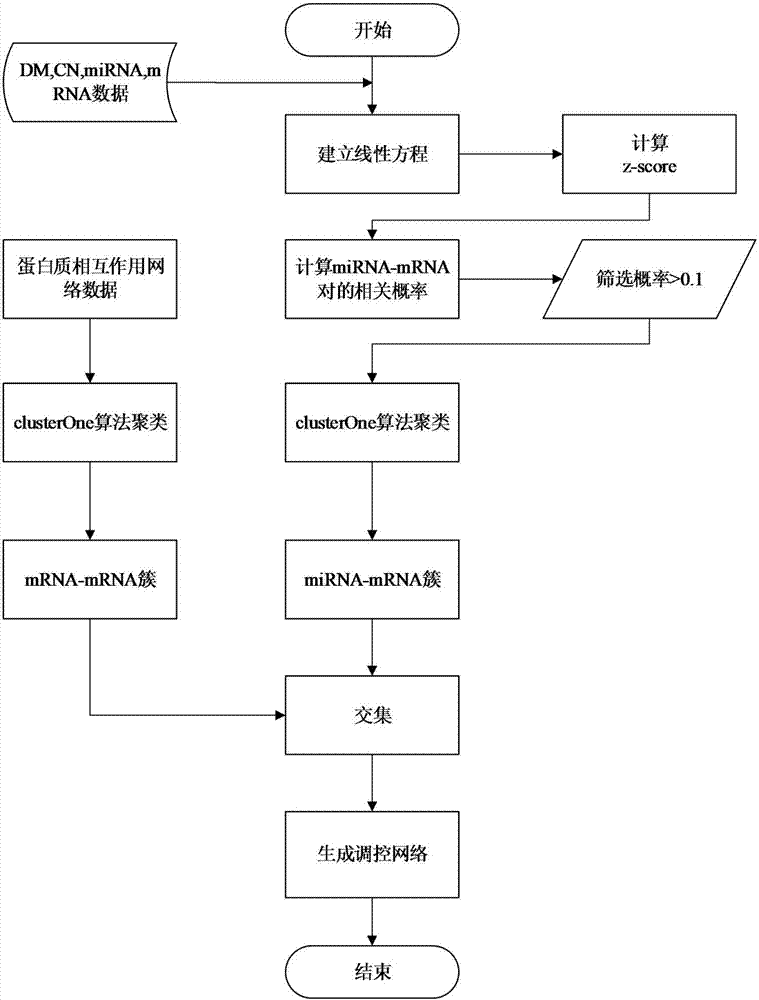
:生物信息学是一门生命科学和计算机科学相结合的新兴学科,生物信息的采集、处理、存储、传播,分析和解释等,通过综合利用生物学、计算机科学和信息技术来揭示复杂的生物数据所蕴藏的生物学奥秘。由于现代技术的提高人们已经能够得到全基因组上基因表达的数据以及目前所发现所有的mirna的数据,并且mirna对于肿瘤发生与发展的重要性也越来越被人们所重视。然而目前对mirna调控网络的研究主要存在以下三大问题:(1)mirna调控网络是一个二分网络,由于mirna之间不存在直接调控作用,而是mirna调控mrna,但是部分mrna可以调控mirna形成反馈调节模块,因此由mirna与靶基因所构成的网络为二分网络。现有方法不是针对二分网络的,很难直接分析得到mirna之间的协同调控作用,使得适用于蛋白质相互作用网络的算法无法直接用于mirna调控网络;(2)由于mirna调控网络关系通常比较复杂,是一种多对多的关系,调控的多样性使得在mirna调控网络中很难找到mirna的调控模块;(3)目前存在大量的生物数据信息,如转录因子调控数据、甲基化数据、拷贝数数据和蛋白质相互作用网络数据等,如何利用这些数据来协助发现mirna调控网络是当前研究面临的一个挑战。对与泛癌症的探讨已经很多成果,然而大多数针对mirna数据以及mrna数据,没有从多角度考虑两者之间的调控关系,比如很多研究者不考虑甲基化,拷贝数等因素对基因表达的影响,从而使得mirna表达与基因表达之间的调控关系并不详尽。目前为止,由于数据过于庞大,很少有人从多角度多因素对泛癌症的mirna表达和基因表达之间的调控关系进行系统全面的详细剖析。本研究在之前的研究基础上,通过合理设定阈值来减少数据量,最终得到实验结果。综上所述,现有技术存在的问题是:没有专门针对二分网络的调控模块发现方法,且没有很好利用现有的多种测序数据。技术实现要素:针对现有技术存在的问题,本发明提供了一种构建mirna调控网络的方法。本发明是这样实现的,一种构建mirna调控网络的方法,所述构建mirna调控网络的方法包括以下步骤:步骤一,应用线性回归计算mrna和mirna的线性回归系数;利用mirna的线性回归系数与其标准差,将mirna与mrna的线性回归系数转化为t统计量;根据t分布,以样本数量d为自由度,计算出z-score;步骤二,通过localfdr算法来计算mirna与mrna的相关概率;以mirna-mrna的相关概率为边的权重建立原始的mirna与mrna的调控网络;步骤三,通过clusterone聚类算法将mirna调控网络聚类,得到独立的mirna-mrna簇;对蛋白质相互作用网络数据通过clusterone算法聚类,得到mrna-mrna簇;对mirna-mrna簇和mrna-mrna簇进行分析,得到两种簇之间的交集;用交集数据对之前得到的原始mirna与mrna调控网络进行优化,生成最终的调控网络。进一步,所述mrna和mirna建立线性回归方程来计算mrna和mirna的相关度,mirna在线性回归方程中的系数数学公式表示为:其中β0是常数项,和分别代表拷贝数的回归系数,dna甲基化的回归系数和mirna的回归系数;其中yi,t,d代表mrnai在疾病d的样本t中的表达值,代表在疾病d的样本t中mirnak对应mrnai的系数。进一步,所述将系数转化为z-score,并做数据筛选:将系数转化为t统计量:其中是系数的标准差;根据t分布、以样本数量d为自由度,计算出z-score,得到的z-score与ti,k,d成正比:zi,k,d代表疾病d中基因i与mirnak的z-score,最终的z-score接近于正态分布,即zi,k,d~n(0,1)。进一步,通过clusterone聚类算法将得到的网络聚类,建立mirna与mrna的调控网络聚类,得到相对独立的mirna-mrna簇;对蛋白质相互作用网络数据通过clusterone算法得到mrna-mrna簇;通过两组数据来找到两种簇之间的交集,通过两种簇之间的交集构建mirna-mrna的调控模块。进一步,clusterone算法主要是建立在内聚力分数基础之上,内聚力分数定义如下:其中win(v)代表节点集合v中所有的边的权重,wbound(v)代表与节点集合v相连接所有的边的权重,p(v)则代表惩罚项。本发明的另一目的在于提供一种应用所述构建mirna调控网络的方法的mirna调控网络。本发明的优点及积极效果为:从多角度多因素探讨泛癌症的mirna表达和基因表达之间的调控关系,保证mirna与mrna调控关系的识别效能,为疾病的生物学病理研究提供基础。本发明综合考虑mirna、dna甲基化和拷贝数对基因表达的影响,能够估计mirna和mrna之间的调控关系,建立mirna调控网络,对复杂疾病的致病机理研究、疾病风险预测和生物药物研制等都有重要的意义。目前运用最多的就是靶向治疗,通过本研究的mirna-mrna匹配对的数据,可进行基因的靶向治疗,也可以为研制靶向治疗的药物提供一定的借鉴。某些肿瘤是由于单一致癌基因的异常激活而形成并依赖于该异常基因的激活,这种现象称为致癌基因依赖。识别可用药的致癌驱动因子创造了可使用高效治疗性干预的可能性。本发明引入dna甲基化数据和拷贝数数据,可以更加精确的估计mirna-mrna的调控关系,从而建立的网络可信度更高,相关证明可参考后面实施例中的结果和结论;靶向治疗是减少癌症死亡率的重要措施之一,而癌症的mirna调控网络的研究可以为靶向治疗等提供依据。而对于泛癌症的mirna调控的调控网络而言,更有可能有些癌症的mirna调控作用在早期就已经发生了,因此能够对早期癌症的诊断和预防发挥作用。另外,mirna的调控网络对于生物通路的构建也有一定的借鉴价值,通过对mirna调控网络的详细分析,可以对关键基因使用靶向治疗调控相应其表达,重新激活癌症的关键信号通路,对于癌症的医治意义重大。附图说明图1是本发明实施例提供的构建mirna调控网络的方法流程图。图2是本发明实施例提供的构建mirna调控网络的方法实现流程图。图3是本发明实施例提供的12种癌症的本地错误发现率示意图。图4是本发明实施例提供的clusterone算法的贪婪策略示意图。图5是本发明实施例提供的在实验2上的实验结果示意图。具体实施方式为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。下面结合附图对本发明的应用原理作详细的描述。如图1所示,本发明实施例提供的构建mirna调控网络的方法包括:s101:应用线性回归计算mrna和mirna的线性回归系数;利用mirna的线性回归系数与其标准差,将mirna与mrna的线性回归系数转化为t统计量;根据t分布,以样本数量d为自由度,计算出z-score;s102:通过localfdr算法来计算mirna与mrna的相关概率;以mirna-mrna的相关概率为边的权重建立原始的mirna与mrna的调控网络;s103:clusterone聚类算法将mirna调控网络聚类,得到独立的mirna-mrna簇;对蛋白质相互作用网络数据通过clusterone算法聚类,得到mrna-mrna簇;对mirna-mrna簇和mrna-mrna簇进行分析,得到两种簇之间的交集;用交集数据对之前得到的原始mirna与mrna调控网络进行优化,生成最终的调控网络。下面结合附图对本发明的应用原理作进一步的描述。如图2所示,本发明实施例提供的构建mirna调控网络的方法的具体实现步骤如下:步骤一,对样本数据进行预处理:如果样本数据是零值或者缺失,则以平均值代替。步骤二,通过线性回归模型对数据进行处理:利用癌症的dna甲基化数据,拷贝数数据,mrna数据和mirna数据建立线性回归方程来计算mrna和mirna的相关度(即mirna在线性回归方程中的系数)。其数学公式可表示为:其中β0是常数项,和分别代表拷贝数的回归系数,dna甲基化的回归系数和mirna的回归系数。其中yi,t,d代表mrnai在疾病d的样本t中的表达值,代表在疾病d的样本t中mirnak对应mrnai的系数。步骤三,将系数转化为z-score,并做数据筛选:首先将系数转化为t统计量:其中是系数的标准差。根据t分布、以样本数量d为自由度,计算出z-score,得到的z-score与ti,k,d成正比:zi,k,d代表疾病d中基因i与mirnak的z-score,最终的z-score接近于正态分布,即zi,k,d~n(0,1)。本发明实施例采用12种癌症的数据,筛选出z-score在9种癌症种类中为负值的mirna-mrna对,以减少计算量。步骤四,计算出mirna与基因的在癌症中的相关概率,并且筛选相关概率大于0.1的mirna-mrna对,建立mirna调控网络。步骤五,通过clusterone聚类算法将得到的网络聚类。先对建立mirna与mrna的调控网络聚类,得到的结果是相对独立的mirna-mrna簇。再对蛋白质相互作用网络数据通过clusterone算法得到mrna-mrna簇。通过这两组数据来找到两种簇之间的交集,通过两种簇之间的交集构建mirna-mrna的调控模块。clusterone算法主要是建立在内聚力分数基础之上,内聚力分数定义如下:其中win(v)代表节点集合v中所有的边的权重,wbound(v)代表与节点集合v相连接所有的边的权重,p(v)则代表惩罚项,加入惩罚项是因为可能存在数据不确定性以及那些在节点集合中未发现的节点。实验中内聚力分数取最大值。下面结合实验对本发明的应用效果作详细的描述。实验1:mirna与mrna在12种癌症中的相关概率的计算模拟数据集有12组z-score数据,每组数据集行数为17788,每组数据集列数为677。行代表mrna,列代表mirna。并且每组数据都是这样的17788*677得矩阵,值为z-score。表1为部分mirna-mrna对的相关概率。表1mirna名称编号基因名概率hsa-let-7b-3pmimat0004482diablo0.104069771hsa-let-7b-5pmimat0000063eral10.232141067hsa-let-7d-3pmimat0004484wisp20.202205556hsa-let-7d-3pmimat0004484c7orf100.13693383hsa-let-7d-5pmimat0000065sema3c0.469382042hsa-let-7g-3pmimat0004584synpo0.715390956hsa-mir-100-5pmimat0000098atp5c10.247348288hsa-mir-101-3pmimat0000099lrrc590.72080575hsa-mir-101-3pmimat0000099ckap40.555390758hsa-mir-101-3pmimat0000099adm0.304017557hsa-mir-101-3pmimat0000099psmd20.159739237hsa-mir-101-5pmimat0004513lrrc590.784917586hsa-mir-103a-3pmimat0000101tgfb1i10.335263029具体实施步骤如下:1、运用localfdr算法来计算mirna与mrna的相关概率。z-score接近于正态分布,即zi,k,d~n(0,1)。其中计算方法为:ti,k,d是一个变量,表明基因j与mirnak是否为匹配对,如果ti,k,d=1表明基因i与mirnak为真匹配对,否则就不是。pd0代表不是匹配对的概率,pd1代表是匹配对的概率。2、对于给定z-score的基因i与mirnak的匹配对,其概率可计算如下:实验2:对mirna调控网络和ppi网络聚类,并找到两个网络的交集。clusterone算法的贪婪策略的步骤描述,该贪婪策略共分为五步。(1)让v0={v0},并且设置t=0;(2)计算vt,并且使vt+1=vt;(3)对每一个至少具有一个边界边(boundaryedge)的内部节点v,计算v'=vt∪{v};(4)对每一个至少具有一个边界边的内部节点v,计算内聚力分数f(v”)=vt-{v},如果f(v”)>f(vt+1),则vt+1=v”。(5)如果vt≠vt+1,则增加t并且返回到步骤(2),否则就称集合vt为最优的子网络。这个子网络的增长过程中,允许去除任何一个节点,包括原始的种子节点。如果最后子网络中不包含原始种子节点,则说明该种子节点对于子网络而言是一个外部节点。下面结合具体应用实施例对本发明的应用原理作进一步的描述。1、假如现在有11个节点,其中有七个节点被标记为a-g。图3为clusterone算法的贪婪策略。假设p(v)=0,则当前的集合v即深色阴影部分的内聚力分数f(v)=10/15,在步骤3中,算法只能通过增加节点c、f和g,在步骤4中,算法只能通过删除a、b、d或e。在步骤3中,最后的选择是假如节点c,因为增加节点c后,不仅将三个边界边a-c、b-c和d-c转化为内部边,而且没有引入任何边界边。在集合v中假如c后,其内聚力分数为f(v)=13/15。加入f节点后内聚力分数f(v)=14/17,加入f节点后内聚力分数f(v)=14/18。2、clusterone算法第二步中定义了一个重叠系数w(a,b),其定义为:如果集合a与b的重叠系数w(a,b)>0.8,则将a和b合并成为一个更大的网络结构。3、利用clusterone算法得到两种类型的网络子结构,取两种网络子结构中有交集的簇构建mirna调控模块。表2为本发明实施例的mirna调控模块。表2实验3:对mirna调控网络的基因做kegg富集分析。对mirna调控网络中的所有基因进行通路富集分析,得到的富集结果如表3所示。其中,每一行数据表明基因富集的生物过程编号、显著性p值、富集基因数、生物过程总基因数、生物过程描述。表3由表3中的keggpathway富集结果可知,研究预测的mirna调控网络中的基因集在26个生物通路中出现显著富集,并且p-value的值都小于0.05。表明mirna调控网络中的mirna的异常表达可能对多个肿瘤相关信号通路中的基因存在影响。泛癌症mirna调控网络中的基因在大多数癌症中相关生物进程中具有关键作用。各类基因主要富集的生物过程有:粘着斑、细胞外基质受体交互、金黄色葡萄球菌感染、吞噬体、扩张型心肌病、肥厚性心肌病、补体和凝血因子的瀑布反应、蛋白质的消化和吸收、病毒性心肌炎、造血细胞谱系、白细胞穿内皮移行、致心律失常性右室心肌病、肌动蛋白细胞骨架调节、肠道免疫网络免疫球蛋白a的生产、细胞粘附分子、利什曼病、疟疾、阿米巴病、癌症通路、破骨细胞分化、溶酶体、弓形体病、细胞因子-细胞因子受体相互作用、哮喘、类风湿性关节炎、转化生长因子-β信号通路等癌症相关生物过程。值得注意的是富集结果中癌症通路(pathwaysincancer)是keggpathway数据库对各种癌症的通路的一个整合。癌症通路中包含了结肠直肠癌(crc)、胰腺癌(pancreascancer)、神经胶质瘤(glioma)、甲状腺腺瘤(thca)、急性髓性白血病(acutemyeloidleukemia)、慢性骨髓性白血病(chronicmyeloidleukemia)、基底细胞癌(basalcellcarcinoma)、黑素瘤(melanomas)、肾细胞癌(renalcellcarcinoma)、膀胱癌(bladdercancer)、前列腺癌(prostatecancer,prad)、子宫内膜癌(endometrialcancer)、小细胞肺癌(smallcelllungcancer)和非小细胞性肺癌(non-smallcelllungcancer)等癌症。其中结肠直肠癌(crc)、甲状腺腺瘤(thca)、前列腺癌(prostatecancer,prad)和肾透明细胞癌(kidneyrenalclearcellcarcinoma,kirc)正是我们所用到的数据集,需要指出的是肾透明细胞癌是肾细胞癌的一种类型。因此,结果也证明了本发明的方法能够找到与癌症直接或间接相关的基因。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种玉米籽粒发育相关蛋白、编码基因及应用的制作方法与工艺
- 一种龙葵花青素合成调控基因SnATCN及其应用的制作方法与工艺
- 一种基于CRISPR‑Cas9的基因调控装置及基因调控方法与流程
- 一种基于电磁诱导透明的群时延调控器的制作方法与工艺
- 一种热应激条件下奶牛乳腺差异表达基因调控网络的构建方法与流程
- 一种基于碳氮流定向调控的混菌发酵“一锅法”生产微生物油脂的方法与流程
- GhPME36基因在调控植物次生壁发育中的应用的制作方法与工艺
- 一种调控蚜虫碳氢化合物合成基因及其应用的制作方法与工艺
- 玉米锌铁调控转运体ZmZIP8基因及其应用的制作方法与工艺
- 拟南芥SSCD1基因突变在调控植物体内茉莉酸合成中的应用的制作方法与工艺