快速减轻帕金森病中的运动波动的制作方法
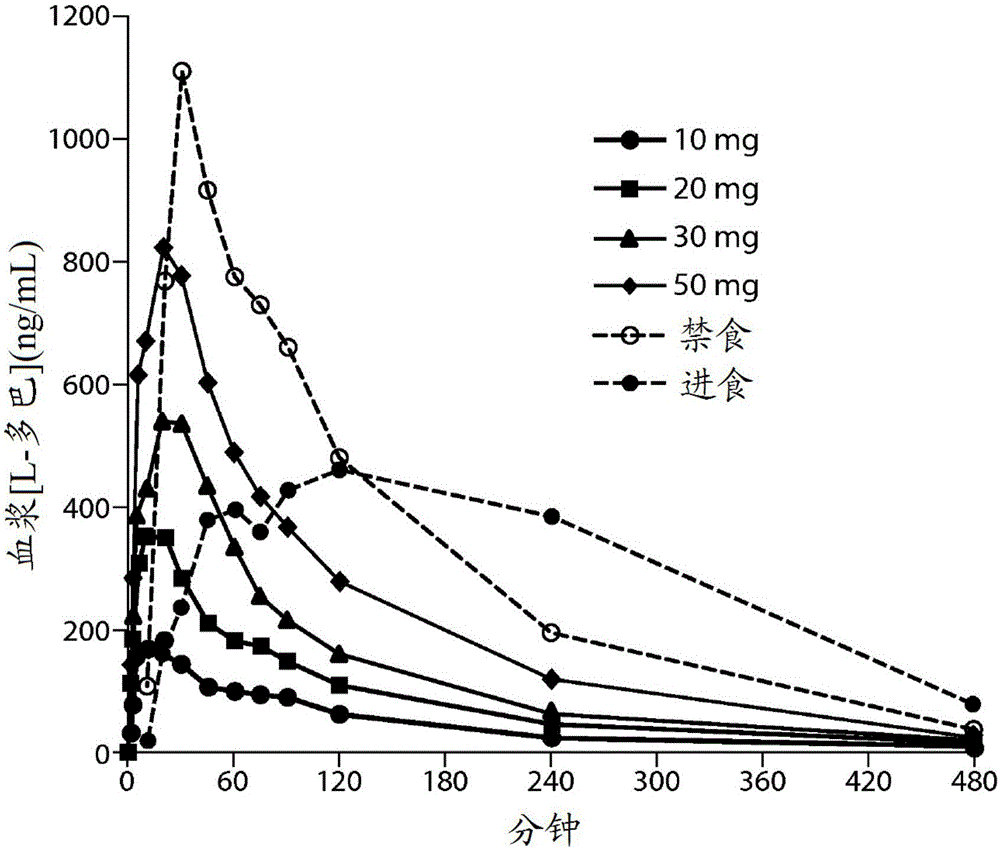
帕金森病(Parkinson’sdisease,在本文中也称为“PD”)以神经病理学上基底神经节中多巴胺神经元的退化和神经学上虚弱性震颤、运动缓慢和平衡问题为特征。据估计有超过一百万人患有帕金森病。几乎所有患者都接受多巴胺前体左旋多巴或“L-多巴”,其通常与多巴脱羧酶抑制剂卡比多巴结合。在疾病的早期阶段,L-多巴足以控制帕金森病的症状。但是,在一段时间以后,其倾向于不太有效,所述一段时间可以为疾病进程的数月到数年不等。L-多巴有效性降低的一个实例是进行治疗的受试者中运动波动(motorfluctuations)的出现。“运动波动”意指受试者开始对多巴胺替代疗法表现出可变的响应,使得在一些时间段内治疗剂表现出良好效力并且充分控制帕金森病的症状(在本文中也称为“ON”时间/发作(episode)或“ON”),而在另一些时间段药剂表现为几乎没有效果并且帕金森病的症状恶化,其在本文中也称为“OFF”时间/发作或“OFF”。运动波动可表现为效力“减退”,L-多巴的效力不再像开始观察的那样长,并且接着发生‘on-off’综合征,其中患者经历运动性的失能波动。逐渐地,经过一段时间,L-多巴的效力(on时间)可降低到多巴胺能治疗的有用性严重受限的程度。认为帕金森病患者中L-多巴变化的效力至少部分地与L-多巴的血浆半衰期有关,即使在与卡比多巴共施用时,所述半衰期也倾向于非常短,在1至3小时的范围。在疾病的早期阶段,该因素被靶纹状体神经元的多巴胺储存能力减轻。L-多巴被所述神经元摄取和储存并且随时间释放。但是,随着疾病进展,多巴胺能神经元退化,导致多巴胺储存能力降低。因此,L-多巴的积极效果变得与L-多巴的血浆水平的波动越来越相关。此外,患者倾向于出现涉及L-多巴的胃排空和差的肠摄取的问题。左旋多巴不稳定的胃排空造成运动性的随机波动。患者表现出帕金森病症状越来越显著的摆动,从血浆水平降低时恢复到典型帕金森疾病症状到L-多巴施用后血浆水平临时升得过高时所谓的运动障碍。需要提供帕金森患者中运动波动和OFF发作的快速减轻,其中所述效果发生在临床有意义的时期,并且其中所述效果允许患者充分的响应持续时间。发明概述本发明提供了用于治疗帕金森病患者中的OFF发作的方法,包括向患者的肺部系统施用左旋多巴,其中在施用后,所述患者的帕金森病统一评定量表(UnifiedParkinson’sDiseaseRatingScale,UPDRS)第3部分(也称为“UPDRS第III部分”或“UDPRSIII”)评分与安慰剂对照相比提高例如至少约5个点,和/或其中在施用后,所述患者的帕金森病统一评定量表(UPDRS)第3部分评分与施用前患者的UDPRS第3部分评分相比提高例如至少约5个点。在一个优选实施方案中,患者的UPDRS第3部分评分在例如施用左旋多巴的约60分钟内提高。本发明还提供了减少平均每日OFF时间的方法和向患者递送左旋多巴的方法。本发明特别可用于在帕金森患者中降低平均每日OFF时间和OFF发作的持续时间。附图简述图1:90/8/2吸入和口服左旋多巴施用后的平均血浆左旋多巴浓度对时间数据。图2:与口服施用相比90/8/2吸入后的平均血浆左旋多巴浓度对时间数据。图3:吸入50mg90/8/2或者在进食和禁食条件下口服施用100mg左旋多巴(CD/LD25/100mg)后个体受试者中的血浆左旋多巴浓度。图4:左旋多巴AUC0-∞对90/8/2细颗粒剂量。图5:左旋多巴Cmax对90/8/2细颗粒剂量。图6:平均血浆浓度的药代动力学建模。符号表示观察到的平均浓度,线表示通过模型预测的浓度。图7:具有和不具有卡比多巴(CD)预治疗的情况下的平均左旋多巴血浆浓度。图8:患者血浆左旋多巴浓度与UPDRS评分相比。图9:是示出了在作为主要终点的第6次访问时接受50mg90/8/2细颗粒剂量的剂量水平2的研究药物的患者和接受安慰剂的患者之间UPDRS第3部分评分对以分钟为单位的时间的平均变化的线图。图10:是示出了在作为主要终点的第4次访问时接受35mg90/8/2细颗粒剂量的剂量水平1的研究药物的患者和接受安慰剂的患者之间UPDRS第3部分评分对以分钟为单位的时间的平均变化的线图。图11:表明具有运动障碍的ON时间没有恶化。图11A示出了90/8/2与安慰剂相比无麻烦的运动障碍(non-troublesomedyskenisia)时间(小时)随着时间(周)的变化。图11B示出了90/8/2与安慰剂相比麻烦的运动障碍(troublesomedyskenisia)时间(小时)随着时间段(周)的变化。发明详述定义半衰期(T1/2)是体液或组织中的药物浓度(C)达到浓度C/2的时间。“CmaxPul”意指在肺部递送后测量的观察到的最大血浆浓度(Cmax)。“Cmaxoral”意指口服递送后测量的观察到的最大血浆浓度。曲线下面积AUC对应于给定时间间隔的血浆浓度的积分。AUC以质量单位(mg,g)×升-1×小时表示,是药物的生物利用度的量度。“AUCPul”意指在肺部递送后测量的血浆浓度对时间曲线下的面积(AUC)。“AUCoral”意指在口服递送后测量的血浆浓度对时间曲线下的面积(AUC)。术语“变差系数”(CV)以%CV表示,定义为标准偏差σ与平均值μ的比:Cv=σ/μ本文使用的语句“标称剂量(nominaldose)”或“标称粉末剂量(nominalpowderdose)”意指容器中包含的颗粒的总质量中存在的左旋多巴的百分比,表示可用来向患者施用的左旋多巴的最大量。“细颗粒分数(fineparticlefraction)”或“FPF”对应于容器中存在的颗粒质量中空气动力学直径小于5.6μm的颗粒的百分比。本文使用的术语“细颗粒剂量”定义为标称剂量乘以FPF。如本文使用的,患者中“平均每日OFF时间的减少”是指在患者日志中记录的或通过临床医生观察的患者每日OFF时间的平均减少。本文使用的帕金森病统一评定量表(UPDRS)是用于测量帕金森病的体征和症状的良好确定的工具。整个UPDRS由四部分组成。第1、2和3部分包括44道问题。除非另有说明,所有项目从零(正常)到四(严重受影响)评定,每个项目用简短语句定义。UPDRS包括通过临床医生(运动检查)以及心理功能和日常生活活动(通过询问患者获得的ADL)的历史报告二者评分。第1部分测量心理状态、行为和情绪,包括智力障碍、思维障碍、积极性/主动性和抑郁症。第2部分测量日常生活活动(ADL),包括讲话、唾液分泌、吞咽、书写、切食物、穿衣服掉落、僵硬行走(freezingwalking)、震颤和感觉主诉(sensorycomplaints)。第3部分是运动检查,测量包括讲话、面部表情、静止时的震颤、活动震颤、僵硬、手指轻拍(fingertaps)、手运动、手旋前和旋后、腿灵活度、从椅子中起立、摆姿势、步态、姿态稳定性和身体运动徐缓(bodybradykinesia)。第4部分测量治疗的并发症,包括运动障碍持续时间、残疾疼痛(disabilitypain)、off时期和持续时间、睡眠障碍等。缩写列表本发明的特征和其他细节将在权利要求书中更具体描述和指出。应理解的是,本发明的具体实施方案仅以举例说明的方式示出并且不限制本发明。本发明的原理特征可以在多个实施方案中使用而不脱离本发明的范围。如本文和所附权利要求书中使用的,单数形式“一”、“一个”和“所述”包括复数指示物,除非上下文明确指出相反情况。根据本发明,本文中使用的术语“左旋多巴的剂量”意指包含一定量的适于通过吸入向患者递送的剂型的左旋多巴的制剂。在一个实施方案中,根据本发明的左旋多巴的剂量包括含左旋多巴的颗粒。用于向呼吸系统递送左旋多巴的颗粒和方法描述在例如美国专利No:6,514,482和美国专利再公布No.RE43711中,两者的内容通过引用整体并入本文。颗粒优选为干粉形式,并且通过细颗粒分数(FPF)、几何尺寸和空气动力学尺寸及其他性质表征,如下文进一步描述的。使用阶式碰撞取样器(Cascadeimpactor)的重量分析是测量空气中颗粒的粒径分布的方法。Andersen阶式碰撞取样器(AndersenCascadeImpactor,ACI)是8平台碰撞取样器,其可以基于空气动力学粒径将气溶胶分离成9种不同级分。每个平台的截留尺寸取决于操作ACI的流速。优选地ACI以60L/min校准。在一个实施方案中,二平台缩减ACI(two-stagecollapsedACI)用于颗粒优化。二平台缩减ACI由八平台ACI的平台0、2和F组成并且允许收集两种分离的粉末级分。在每个平台,气溶胶流通过喷嘴并且撞击在表面。气溶胶流中具有足够大惯性的颗粒将撞击在板上。不具有足够惯性来撞击板的较小颗粒将保留在气溶胶流中并且别携带到下一平台。校准ACI以使得第一平台上收集的级分被称为细颗粒级分FPF(5.6)。该FPF对应于空气动力学直径小于5.6μm的颗粒的%。通过ACI的第一平台并且沉积在收集滤器上的粉末级分被称为FPF(3.4)。这对应于空气动力学直径小于3.4μm的颗粒的%。FPF(5.6)级分已被证明与沉积在患者肺中的粉末级分相关,而FPF(3.4)已被证明与到达患者深肺的颗粒级分相关。本发明至少50%颗粒的FPF小于约5.6μm。例如,但不限于,至少60%、或70%、或80%、或90%颗粒的FPF小于约5.6μm。测量空气中颗粒(airborneparticle)的粒径分布的另一种方法是多平台液体碰撞取样器(multi-stageliquidimpinger,MSLI)。多平台液体碰撞取样器(MSLI)以与Anderson阶式碰撞取样器(ACI)相同的原理操作,但不是八个平台,MSLI中具有五个平台。此外,并不是每个平台由固体板构成,每个MSLI平台由甲醇润湿的玻璃熔块构成。润湿的平台用于阻止弹跳和再夹带,这些情况在使用ACI时可能发生。MSLI用于提供粉末流速依赖性的指示。这可以通过以30、60和90L/min操作MSLI并且测量收集在平台1和收集滤器上的粉末级分来实现。如果不同流速下每个平台上的级分保持相对恒定,则认为粉末接近不依赖流速。本发明颗粒的振实密度小于约0.4g/cm3。振实密度小于约0.4g/cm3的颗粒在本文中称为“空气动力学轻颗粒(aerodynamicallylightparticles)”。例如,颗粒的振实密度小于约0.3g/cm3、或振实密度小于约0.2g/cm3、振实密度小于约0.1g/cm3。可使用本领域技术人员已知的装置测量振实密度,例如双平台微处理器控制的振实密度测试仪(Vankel,NC)或GEOPYCTM装置(MicrometriesInstrumentCorp.,Norcross,GA30093)。振实密度是包络质量密度(envelopemassdensity)的标准量度。可使用以下文献中的方法测量振实密度:USPBulkDensityandTappedDensity,UnitedStatesPharmacopiaconvention,Rockville,MD,第10次增补版,4950-4951,1999。可有助于低振实密度的特征包括不规则表面纹理和多孔结构。各向同性颗粒的包络质量密度定义为颗粒的质量除以可以将其包围在里面的最小球体包络体积。在本发明的一个实施方案中,颗粒的包络质量密度小于约0.4g/cm3。本发明颗粒具有优选尺寸,例如,至少约1微米(μm)的体积中值几何直径(volumemediangeometricdiameter,VMGD)。在一个实施方案中,VMGD为约1μm至30μm,或由约1μm至30μm涵盖的任何子范围,例如,但不限于,约5μm至约30μm、或约10μm至30μm。例如,颗粒的VMGD为约1μm至10μm、或约3μm至7μm、或约5μm至15μm、或约9μm至约30μm。颗粒的中值直径、质量中值直径(massmediandiameter,MMD)、质量中值包络直径(massmedianenvelopediameter,MMED)或质量中值几何直径(massmediangeometricdiameter,MMGD)为至少1μm,例如,5μm或者接近或大于约10μm。例如,颗粒的MMGD大于约1μm并且至约30μm,或者约1μm至30μm涵盖的任何子范围,例如,但不限于,约5μm至30μm,或者约10μm至约30μm。喷雾干燥颗粒的直径(例如,VMGD)可以使用激光衍射仪器(例如,Helos,由Sympatec,Princeton,NJ制造)测量。用于测量颗粒直径的其他仪器是本领域中周知的。样品中颗粒直径的范围取决于因素如颗粒组成和合成方法。可以选择样品中颗粒的粒径分布以最佳地沉积在呼吸道内的靶部位。空气动力学轻颗粒的“质量中值空气动力学直径”(MMAD)(在本文中也称为“空气动力学直径”优选地为约1μm至约5μm,或者由约1μm至约5μm涵盖的任何子范围。例如,MMAD为约1μm至约3μm,或者MMAD为约3μm至约5μm。在实验中,可以通过使用重力沉降法测定空气动力学直径,其中将全部颗粒沉降一定距离的时间用于直接推断颗粒的空气动力学直径。用于测量质量中值空气动力学直径(MMAD)的一种间接方法是多平台液体碰撞取样器(MSLI)。可以由以下方程式估计空气动力学直径daer:daer=dg√ρtap其中dg是几何直径,例如MMGD,ρ是粉末密度。振实密度小于约0.4g/cm3,中值直径为至少约1μm,例如至少约5μm,空气动力学直径为约1μm至约5μm,优选约1μm至约3μm的颗粒更加能够逃脱口咽区的惯性和重力沉积,并靶向气道,特别是深肺。使用较大、更多孔的颗粒是有利的,因为其比较小、致密的气溶胶颗粒(例如目前用于吸入疗法的那些)更有效气溶胶化。与较小、相对致密的颗粒相比,较大的空气动力学轻颗粒(优选中值直径为至少约5μm)可潜在地更成功避免肺泡巨噬细胞的吞噬和从肺部清除,这是因为其尺寸将颗粒排除在吞噬细胞胞质空间之外。随着颗粒直径增大超过约3μm,肺泡巨噬细胞对颗粒的吞噬急剧降低。Kawaguchi,H.,等,Biomaterials,7:61-66(1986);Krenis,L.J.和Strauss,B.,Proc.Soc.Exp.Med.,107:748-750(1961);以及Rudt,S.和Muller,R.H.,JContr.Rel.,22:263-272(1992)。对于统计学上各向同性形状的颗粒,例如具有粗糙表面的球体,颗粒包络体积约等于巨噬细胞内用于完成颗粒吞噬所需的胞质空间的体积。可以制备具有合适的材料、表面粗糙度、直径和振实密度的颗粒,以局部递送至呼吸道的选择区域,例如深肺或者上或中央气道。例如,较高密度或较大颗粒可用于上气道递送,或者样品中提供有相同或不同治疗剂的不同尺寸颗粒的混合物可在一次施用中施用以靶向肺中的不同区域。对于中央和上气道递送,空气动力学直径为约3至约5μm的颗粒是优选的。对于深肺递送,空气动力学直径为约1至约3μm的颗粒是优选的。在正常呼吸条件下,气溶胶的惯性碰撞和重力沉积是气道和肺腺泡中的主要沉积机制。Edwards,D.A.,J.AerosolSci.,26:293-317(1995)。两种沉积机制的重要性与气溶胶的质量成比例增加,而不是颗粒(或包络)体积。由于气溶胶在肺中的沉积部位由气溶胶的质量决定(至少对于平均空气动力学直径大于约1μm的颗粒),通过增加颗粒表面不均匀性和颗粒多孔性降低振实密度允许将所有其他物理参数相等的较大颗粒包络体积递送到肺部。与实际包络球体直径相比,低振实密度颗粒具有小的空气动力学直径。空气动力学直径daer与包络球体直径d通过以下简化公式相关(Gonda,I.,"Physico-chemicalprinciplesinaerosoldelivery,"于TopicsinPharmaceuticalSciences1991(编辑D.J.A.Crommelin和K.K.Midha)中,第95-117页,Stuttgart:MedpharmScientificPublishers,1992)):daer=d√ρ其中包络质量密度的单位是g/cm3。对于约daer=3μm的空气动力学直径,在人肺的肺泡区中发生单分散气溶胶颗粒的最大沉积(~60%)。Heyder,J.等,J.AerosolSci.,17:811-825(1986)。由于其较小的包络质量密度,包含将表现出最大深肺沉积的单分散吸入粉末的空气动力学轻颗粒的实际直径为:d=3/√ρμm(其中ρ_<1g/cm3);其中d始终大于3μm。例如,对于包络直径大至9.5μm的颗粒,包络质量密度μ=0.1g/cm3的空气动力学轻颗粒将表现出最大沉积。增加粒径减小了颗粒之间的粘附力。Visser,J.,PowderTechnology,58:1-10。因此,除了有助于较低的吞噬损失以外,大粒径提高了低包络质量密度颗粒气溶胶化至深肺的效率。可以计算空气动力学直径以提供在肺内的最大沉积。之前这通过使用直径小于约5微米,优选约1至约3微米的极小颗粒来实现,其将被吞噬。选择具有较大直径但是足够轻(因此特征描述“空气动力学轻”)的颗粒使得同样递送至肺,但是较大尺寸颗粒不被吞噬。相对于光滑表面,通过使用具有粗糙或不均匀表面的颗粒可以获得改善的递送。在本发明的另一个方面,颗粒的包络质量密度(在本文中也称为“质量密度”)小于约0.4g/cm3。在一些实施方案中,颗粒密度为约0.01、0.02、0.03、0.04、0.05、0.06、0.07、0.08、0.09、小于0.1、0.02至0.05、0.02至0.06g/cm3。质量密度,以及质量密度、平均直径和空气动力学直径之间的关系在2001年7月3日公布的Edwards等的美国专利No.6,254,854中有讨论,其通过引用整体并入本文。可以通过多种方法产生具有上述组成和空气动力学特性的颗粒,包括但不限于喷雾干燥。通常,喷雾干燥技术例如由K.Masters描述在"SprayDryingHandbook",JohnWiley&Sons,NewYork,1984中。本文使用的术语“有效量”或“治疗有效量”意指实现期望效果或效力所需的量。药物的实际有效量可根据所使用的具体药物或其组合、配制的具体组成、施用方式,患者的年龄、体重、病症,以及被治疗发作的严重程度而变化。在多巴胺前体、其激动剂或组合的情况下,其为降低需要治疗的帕金森的症状的量。用于特定患者的剂量在本文中有描述,并且可以由本领域技术人员使用常规考虑(例如,通过合适的常规药理学方案)来决定。可以通过诸如本领域中已知的装置向呼吸系统施用颗粒。例如,从吸入装置如干粉吸入器(DPI)递送颗粒。也可使用计量剂量吸入器(Metered-dose-inhaler,MDI)、喷雾器或滴注技术。在一个实施方案中,通过以下文献中所述的方法将颗粒递送到肺部系统:题目为“HighEfficientDeliveryofaLargeTherapeuticMassAerosol”的美国专利6,858,199和题目为“HighlyEfficientDeliveryofaLargeTherapeuticMassAerosol”的美国专利7,556,798。这两篇专利的整个内容均通过引用并入本文。如其中公开的,将颗粒容纳、包含、储存或封闭在容器(receptacle)中。容器,例如胶囊(capsule)或泡罩(blister)的体积为至少约0.37cm3,并且可具有适于在干粉吸入器中使用的设计。也可以使用体积为至少约0.48cm3、0.67cm3或0.95cm3的较大容器。本文使用的术语“容器”包括但不限于例如胶囊、泡罩、膜覆盖的容器孔(filmcoveredcontainerwell)、区室(chamber)以及本领域技术人员已知在吸入装置中储存颗粒、粉末或可呼吸组合物的其他合适的装置。在一个实施方案中,容器是胶囊,例如标记有特定胶囊尺寸,例如2、1、0、00或000的胶囊。合适的胶囊可以从例如Shionogi(Rockville,MD)获得。在一个实施方案中,胶囊壳可包含羟丙基甲基纤维素(HPMC)。在另一个实施方案中,胶囊壳可包含羟丙基甲基纤维素(HPMC)和二氧化钛。泡罩可从例如HueckFoils,(Wall,NJ)获得。其他适合于在本发明中使用的容器及其体积是本领域技术人员已知的。在一个实施方案中,本发明提供了以少量步骤,优选以单个呼吸活化步骤向肺部系统施用L-多巴。在一个实施方案中,在单个呼吸活化步骤中将储存在吸入器容器中的颗粒质量的至少50%递送到受试者的呼吸系统。在一个实施方案中,在单个呼吸活化步骤中将储存在吸入器容器中的颗粒质量的至少60%,优选至少70%,优选至少80%递送到受试者的呼吸系统。在另一个实施方案中,对于封闭在容器中的颗粒,通过单次呼吸施用将至少1至80毫克L-多巴递送到受试者的呼吸道中。优选地,也可以递送至少10、15、20、25、30、35、40、50、60、75和80毫克。通过使用以相对低的能量如通常由受试者的吸气提供的能量分散的颗粒,增强了在单个呼吸驱动步骤中递送到肺部系统的颗粒。这样的能量在本文中称为“低的”。如本文使用的,“低能量施用”是指这样的施用,其中施加以分散和/或吸入颗粒的能量在吸气期间受试者通常提供的范围内。本发明还涉及向肺部系统有效递送粉末颗粒的方法。例如,但不限于,实际递送标称粉末剂量的至少约60%、优选至少约70%或优选至少约80%。在一个实施方案中,本发明中使用的组合物包含适于肺部递送的颗粒,例如干粉颗粒,其包含按重量(干重)计约60-99%的左旋多巴。特别优选的是包含按重量计约75%或更多左旋多巴的颗粒,甚至更优选包含按重量计约90%或更多左旋多巴的颗粒。颗粒可全部由L-多巴构成,或者还可包含一种或多种额外组分。这些合适的额外组分的实例包括但不限于磷脂、氨基酸、糖和盐。磷脂的具体实例包括但不限于磷脂酰胆碱二棕榈酰磷脂酰胆碱(DPPC)、二棕榈酰磷脂酰乙醇胺(DPPE)、二硬脂酰磷脂酰胆碱(DSPC)、二棕榈酰磷脂酰甘油(DPPG)或其任意组合。本发明颗粒中磷脂如DPPC的量一般小于10wt%。盐包括少量强电解质盐,例如但不限于氯化钠(NaCl)。其他可使用的盐包括柠檬酸钠、乳酸钠、磷酸钠、氟化钠、硫酸钠和碳酸钙。通常,颗粒中存在的盐的量小于10wt%,例如小于5wt%。在一个优选实施方案中,适于通过吸入向患者肺部递送的左旋多巴的制剂包含按重量计90%的左旋多巴、按重量计8%的二棕榈酰磷脂酰胆碱(DPPC)和按重量计2%的氯化钠,其在本文中也称为“90/8/2”。在一个实施方案中,本发明提供了治疗帕金森病患者中的OFF期的方法,包括向患者的肺部系统施用左旋多巴,其中在施用后,所述患者的帕金森病统一评定量表(UPDRS)第3部分评分与安慰剂对照相比提高例如至少约5个点。在一个实施方案中,患者UPDRSIII评分与安慰剂对照相比提高例如至少约8个点,优选至少约10个点,以及优选至少约12个点。在一个实施方案中,向患者施用约30至约60mg细颗粒剂量(fineparticledose,FPD)的左旋多巴,优选90/8/2FPD的左旋多巴。在一个优选实施方案中,向患者的肺部系统施用35mgFPD的左旋多巴。在另一个实施方案中,向患者施用50mgFPD的左旋多巴。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍(dyskinesis)水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,在出现OFF症状时施用FPD的左旋多巴。在一个实施方案中,本发明提供了治疗帕金森病患者中的OFF期的方法,包括向患者的肺部系统施用左旋多巴,其中在施用后,患者的帕金森病统一评定量表(UPDRS)第3部分评分与肺部施用前患者的UPDRSIII评分相比提高例如至少约5个点。在一个实施方案中,患者UPDRSIII评分与安慰剂对照相比提高例如至少约8个点,优选至少约10个点,以及优选至少约12个点。在一个实施方案中,向患者施用约30至约60mg细颗粒剂量(FPD)的左旋多巴,优选90/8/2FPD的左旋多巴。在一个优选实施方案中,向患者的肺部系统施用35mgFPD的左旋多巴。在另一个实施方案中,向患者施用50mgFPD的左旋多巴。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,在出现OFF症状时施用FPD的左旋多巴。在一个实施方案中,本发明提供了用于减少帕金森病患者中平均每日OFF时间的方法,包括一天至少一次,优选一天至少两次向患者的肺部系统施用左旋多巴,其中在施用后,患者的平均每日OFF时间减少至少约1小时,优选至少约2小时,优选至少约3小时,优选至少约4小时,优选至少约5小时或更多。在一个实施方案中,向患者施用约30至约60mg细颗粒剂量(FPD)的左旋多巴,优选90/8/2FPD的左旋多巴。在一个优选实施方案中,向患者的肺部系统施用35mgFPD的左旋多巴。在另一个实施方案中,向患者施用50mgFPD的左旋多巴。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,在出现OFF症状时施用FPD的左旋多巴。在一个实施方案中,本发明提供了向帕金森病患者递送左旋多巴的方法,包括向患者的肺部系统施用左旋多巴,其中在施用后,患者的帕金森病统一评定量表(UPDRS)第3部分评分与肺部施用所述左旋多巴之前患者的UPDRSIII评分相比提高至少约5至约12个点。在一个实施方案中,向患者施用约30至约60mg细颗粒剂量(FPD)的左旋多巴,优选90/8/2FPD的左旋多巴。在一个优选实施方案中,向患者的肺部系统施用35mgFPD的左旋多巴。在另一个实施方案中,向患者施用50mgFPD的左旋多巴。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,在出现OFF症状时施用FPD的左旋多巴。在一个实施方案中,在肺部施用左旋多巴之后,患者的UPDRS第3部分评分与安慰剂对照相比提高至少约5至约15个点,优选至少约5至约12个点,优选至少约5至约10个点,优选至少约5至约8个点。在一个实施方案中,在肺部施用左旋多巴之后,患者的UPDRS第3部分评分与安慰剂对照相比提高至少约2至约15个点,优选至少约2至约12个点,优选至少约2至约10个点,优选至少约2至约8个点,优选至少约2至约5个点,优选至少约3至约15个点,优选至少约3至约12个点,优选至少约3至约10个点,优选至少约3至约8个点,优选至少约3至约5个点,优选至少约4至约15个点,优选至少约4至约12个点,优选至少约4至约10个点,以及优选至少约4至约8个点。在一个实施方案中,在肺部施用左旋多巴后约60分钟内,优选施用后约30分钟内,优选施用后约20分钟内,以及优选施用后约10分钟内,患者的UPDRS第3部分评分与安慰剂对照相比提高。在一个实施方案中,在施用左旋多巴后约60分钟内,优选约30分钟内,优选约20分钟内,以及优选约10分钟内,患者的UPDRS第3部分评分与安慰剂对照相比提高至少约2个点,优选至少约5个点,优选至少约8个点,优选至少约10个点,优选至少约12个点,以及优选至少约15个点。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,与肺部施用左旋多巴之前患者的UPDRS第3部分评分相比,施用后患者的UPDRS第3部分评分提高至少2个点,优选至少约3个点,优选至少约4个点,优选至少约5个点,优选至少约6个点,优选至少约7个点,优选至少约8个点,优选至少约9个点,优选至少约10个点,优选至少约11个点,优选至少约12个点,优选至少约13个点,优选至少约14个点,优选至少约15个点。在一个实施方案中,与肺部施用所述左旋多巴之前患者的UPDRSIII评分相比,患者的UPDRS第3部分评分在施用左旋多巴后约60分钟内,优选约30分钟内,优选约20分钟内,以及优选约10分钟内提高。在一个实施方案中,与肺部施用左旋多巴之前患者的UPDRSIII评分相比,患者的UPDRS第3部分评分在施用左旋多巴后约60分钟内,优选约30分钟内,优选约20分钟内,以及优选约10分钟内提高至少约2个点,优选至少约5个点,优选至少约8个点,优选至少约10个点,优选至少约12个点,以及优选至少约15个点。在一个实施方案中,与肺部施用左旋多巴之前的运动障碍水平相比,患者不经历增加的运动障碍。在一个实施方案中,施用左旋多巴之前患者具有一天约3至约4次OFF发作。在一个实施方案中,施用左旋多巴之前患者具有一天约4至约8小时的OFF发作。在一个实施方案中,在出现OFF症状时施用FPD的左旋多巴。在一个实施方案中,通过吸入向患者施用含有所述FPD的左旋多巴的至少一个胶囊的内容物。在一个实施方案中,通过吸入向患者施用含有所述FPD的左旋多巴,优选90/8/2FPD的左旋多巴的至少两个胶囊的内容物。在一个实施方案中,通过吸入装置将细颗粒剂量的左旋多巴从至少一个胶囊递送至肺部系统。在一个是实施方案中,吸入装置是干粉吸入器(DPI)或计量剂量吸入器(MDI)。在一个实施方案中,本发明方法提供了快速减轻帕金森病患者中的运动波动。本发明方法特别适用于治疗患者中因差的左旋多巴血浆水平控制导致的运动波动。在一个实施方案中,本发明方法包括通过以治疗有效浓度吸入来肺部施用左旋多巴,使得与吸入左旋多巴之前患者血浆中的左旋多巴浓度相比,患者的血浆左旋多巴浓度在吸入后的约10分钟或更短时间内升高至少约200ng/ml,并且其中患者血浆浓度在吸入后至少约15分钟的时期保持升高至少约200ng/ml。在一个实施方案中,患者的血浆左旋多巴浓度在施用后至少约20分钟的时期保持升高至少约200ng/ml。在一个实施方案中,患者的血浆左旋多巴浓度在施用后至少约30分钟的时期保持所述升高至少约200ng/ml。在一个实施方案中,患者的血浆左旋多巴浓度在施用后至少约60分钟的时期保持所述升高至少约200ng/ml。在另一些实施方案中,升高大于200ng/ml、200至500ng/ml、300至400ng/ml、或250至450ng/ml。在一个实施方案中,患者的血浆左旋多巴浓度在10分钟内不会升高大于1000ng/ml。在一个实施方案中,本发明方法提供了快速减轻帕金森病患者中的运动波动,包括通过吸入向患者施用约20mg至约75mg左旋多巴,其中所述患者在所述吸入的10分钟内运动波动得到立即减轻,并且其中所述患者保持所述减轻至少约30分钟的时期。根据本发明的任何方法,与通过吸入施用左旋多巴之前患者的血浆左旋多巴浓度相比,在通过吸入施用一定剂量的左旋多巴后约10分钟时,每4mg施用的左旋多巴使患者血浆中左旋多巴的曲线下面积(AUC)提高至少约1000ng-min/ml。在一个实施方案中,与通过吸入施用左旋多巴之前患者的血浆左旋多巴浓度相比,在通过吸入施用一定剂量的左旋多巴后约10分钟时,每4mg施用的左旋多巴使血浆中所述左旋多巴的AUC提高至少约1000-1500ng-min/ml。根据本发明的任何方法,与通过吸入施用左旋多巴之前患者的血浆左旋多巴浓度相比,在通过吸入施用一定剂量的左旋多巴的约10分钟内,每10mg递送的左旋多巴使患者血浆左旋多巴浓度提高至少约175ng/ml,其中所述患者的血浆左旋多巴浓度在施用后至少约15分钟、优选约20分钟、优选约25分钟、优选约30分钟、优选约45分钟、或优选约60分钟的时期保持至少约175ng/ml的所述升高。在一个实施方案中,本发明提供了快速减轻帕金森病患者中的运动波动的方法,包括通过吸入向患者施用约20mg至约75mg左旋多巴,其中CmaxPul/AUCPul除以CmaxOral/AUCOral大于1,其中口服给予的左旋多巴的剂量与通过肺部递送给予的剂量相对相同。在一个实施方案中,本发明提供了快速减轻帕金森病患者中的运动波动的方法,包括通过吸入施用一个或多个剂量的左旋多巴,其中T1/2/Tmax的比小于1/2,优选小于1/5。在一个实施方案中,本发明任何方法中使用的剂量包括向患者递送约10mg至约75mg左旋多巴。在一个实施方案中,剂量包括约12mg至约35mg左旋多巴。在一个实施方案中,左旋多巴的剂量包括至少约10mg左旋多巴,优选至少约25mg左旋多巴,优选至少约35mg左旋多巴,优选至少约50mg左旋多巴,以及优选至少约75mg左旋多巴。在一个实施方案中,吸入一个或多个胶囊后,向肺部系统递送的左旋多巴的量为约25至约60mg左旋多巴。在另一个实施方案中,吸入一个或多个胶囊后,向肺部系统递送的左旋多巴的量为约35至55mg、约30至50mg、约40至50mg、约45至55mg。在一个实施方案中,本发明任何方法中使用的左旋多巴的剂量包含约30mg至约60mgFPD的左旋多巴。在一个实施方案中,本发明任何方法中使用的剂量为约35mgFPD的左旋多巴。在一个实施方案中,本发明任何方法中使用的剂量为约50mgFPD的左旋多巴。在一些实施方案中,吸入一个左旋多巴胶囊中的粉末后,发生快速的运动减轻或左旋多巴血浆增加。在一些实施方案中,在吸入2、3、4或5个胶囊的粉末后发生快速运动减轻或左旋多巴血浆增加。在一个实施方案中,本发明的任何方法中使用的剂量包含盐。在一个实施方案中,剂量包含磷脂。在一个实施方案中,本发明的任何方法还包括向患者共施用多巴脱羧酶抑制剂。在一个实施方案中,在通过吸入施用左旋多巴之前、与通过吸入施用左旋多巴同时或在通过吸入施用左旋多巴之后,向患者施用多巴脱羧酶抑制剂。在一个实施方案中,本发明的任何方法还可包括向所述患者施用口服剂量的左旋多巴。在一个实施方案中,本发明的任何方法包括保持运动波动减轻至少2小时,优选至少3小时,优选至少4小时,优选至少5小时以及更优选至少6小时或更久。在一个实施方案中,根据本发明的任何方法治疗的帕金森病患者是帕金森病2、3或4期患者。根据本发明的任何方法,左旋多巴的剂量不受中枢神经系统食物效果的影响。在一个优选实施方案中,本发明任何方法中使用的左旋多巴的剂量包含按重量计90%的左旋多巴,按重量计8%的二棕榈酰磷脂酰胆碱(DPPC)和按重量计2%的氯化钠。在根据本发明通过吸入施用左旋多巴的时候,可以同时或顺序地提供超过一种多巴胺前体、DOPA脱羧酶抑制剂或其组合的施用,包括但不限于L-多巴、卡比多巴、阿扑吗啡和苄丝肼。在一个实施方案中,超过一种多巴胺前体或DOPA脱羧酶抑制剂的施用可通过肌肉内、皮下、口服等施用途径来施用。在一些实施方案中,这些其他药剂也通过肺部系统共施用。这些化合物或组合物可在通过吸入肺部施用左旋多巴之前、之后或同时施用,并且在与根据本发明所述方法通过吸入施用的左旋多巴相结合时,认为是“共施用”。在一个实施方案中,患者不需要共施用DOPA脱羧酶抑制剂或允许较低或较少频率剂量的DOPA脱羧酶抑制剂。在另一个实施方案中,患者不需要共施用卡比多巴或者与口服接受L-多巴的患者相比允许较低或较少频率剂量的卡比多巴。在另一个实施方案中,患者不需要共施用苄丝肼或者与口服接受L-多巴的患者相比允许较低或较少频率剂量的苄丝肼。在一个实施方案中,在通过肺部途径施用的左旋多巴和通过口服途径施用的左旋多巴之间,卡比多巴依赖性之间的关系为:其中“w/oCD”意指不具有卡比多巴,“w/CD”意指具有卡比多吧,“INN”指肺部途径,oral指向患者递送卡比多巴的口服途径。在一个实施方案中,需要精确的左旋多巴剂量以使患者为on。例如,在一个实施方案中,左旋多巴的剂量必须使患者的血浆左旋多巴浓度升高约200ng/ml至500ng/ml。有趣的是,左旋多巴浓度的这种小的升高适用于大范围的患者给药方案。可能需要1500-2000ng/ml血浆左旋多巴水平来为“on”的患者可通过血浆中200-500ng/ml的左旋多巴来成为on,而需要500-1000ng/ml血浆左旋多巴水平来为“on”的患者可通过血浆中200-500ng/ml的左旋多巴来成为on。更特别地,可通过使患者血浆浓度升高200-400ng/ml、250-450ng/ml、300-400ng/ml或约375-425ng/ml来使患者为on。可以通过多种方法使患者血浆浓度升高200-500ng/ml。可以口服、通过肺部途径或胃肠外给予左旋多巴。如果通过肺部途径给予,可以向患者的肺部系统提供25-50mg剂量的左旋多巴。在一个实施方案中,向患者肺部系统提供的剂量可以为25-35mg、27-32mg、28-32mg、29-31mg或约30mg。向患者肺部系统提供剂量可以通过多种方式进行。在一个实施方案中,胶囊包含35-40mg左旋多巴粉末,所述胶囊向患者的肺部系统提供胶囊中40-60%的粉末,并且所述粉末包含75-98%左旋多巴。以下实施例旨在举例说明本发明,而不应解释为限制其范围。实施例1概要提供90/8/2干粉左旋多巴制剂,以在成年健康志愿者中评估与口服左旋多巴相比施用90/8/2肺部左旋多巴粉末后的安全性、耐受性和左旋多巴药代动力学(PK)。这些实施例中所述的肺部左旋多巴粉末包含90%左旋多巴、8%二棕榈酰磷脂酰胆碱和2%氯化钠(全部按干重计)的颗粒,并且在本文中称为90/8/2。该数据提供了对单吸入剂量的90/8/2之后左旋多巴的PK的描述,以及与禁食或进食条件下口服施用左旋多巴(LD)的比较,以及具有和不具有卡比多巴(CD)预治疗的PK的比较。这是如下在健康成年男性和女性受试者中进行的二部分研究:部分A-与口服左旋多巴相比的剂量递增阶段;部分B-90/8/2加或减卡比多巴预治疗阶段。部分A是开放标记、3阶段交叉、单递增剂量研究。每位受试者在一个时期(session)在进食或禁食状态下接受单口服剂量的CD/LD(25/100mg),在两个不同的治疗时期(treatmentsession)接受单递增剂量的两种不同剂量的吸入90/8/2(10和30mg或20和50mg左旋多巴细颗粒剂量(FPD))。登记两组,每组9位受试者。部分B是开放标记、随机化、二阶段、阶段平衡的交叉研究。在具有和不具有CD预治疗的情况下,对8位受试者评估单吸入90/8/2剂量(40mg左旋多巴FPD)施用后的安全性、耐受性和左旋多巴PK。在24小时期间收集血样并且使用经验证的液相色谱-串联质谱(LC-MS-MS)测定来确定血浆左旋多巴浓度,所述测定的定量下限为9.84ng/mL。使用非区室方法(non-compartmentalmethod)接着使用具有滞后时间的二区室模型的PK建模来进行药代动力学分析。通过吸入以10至50mg左旋多巴FPD的剂量施用的90/8/2产生了快速升高的与剂量成比例的血浆左旋多巴浓度,在健康成年人中20至50mg的细颗粒剂量后5至10分钟内实现了潜在的治疗相关水平。90/8/2吸入后的左旋多巴血浆浓度比禁食状态下口服施用后的那些升高的更快,比在进食状态下那些快得多。药物施用后最初10分钟的暴露表示为0至10分钟的血浆浓度对时间曲线下的部分面积AUC(AUC0-10m),给药后最初10分钟观察的最大血浆浓度(Cmax,10m)显示与口服施用相比90/8/2吸入后早得多的全身性暴露。与口服施用和对于肺部施用所预期的相比,吸入后受试者与受试者之间的血浆浓度变化性极大降低。分析还表明禁食状态下的口服施用导致与进食状态相比更快速的吸收,但是依然比吸入后慢得多。药代动力学建模显示了与90/8/2吸入后小于0.5分钟的滞后时间相比,进食或禁食状态下口服施用后约9至10分钟的滞后时间。此外,与口服施用相比,吸入后的吸收半衰期更短。在90/8/2吸入后,全身性左旋多巴暴露与施用的90/8/2剂量成比例。施用的90/8/2剂量之间剂量归一化的Cmax和AUC非常类似。与口服施用相比,吸入后剂量归一化的(基于估计的细颗粒剂量)暴露基于AUC大1.3至1.6倍,基于Cmax大1.6至2.9倍。如文献中已经描述的,在口服施用后,在进食受试者中观察到了相当大的Cmax降低和Tmax延长;但是,进食和禁食受试者之间的AUC类似。在交叉设计中具有或不具有卡比多巴预治疗的情况下,来自吸入40mg细颗粒剂量的90/8/2之研究的部分B的血浆浓度显示快速吸收和达到潜在治疗水平的血浆浓度。在没有CD预治疗的情况下,血浆左旋多巴清除快约四倍。相应地,在没有CD预治疗的情况下,Cmax和AUC更低并且Tmax和T1/2稍短。本研究的主要发现是:·吸入90/8/2导致血浆左旋多巴浓度快速升高;·与口服药物施用相比,利用90/8/2吸入给药后最初10分钟基于Cmax和AUC的全身性左旋多巴暴露高得多;·在健康成人中,吸入20至50mg细颗粒剂量后的5至10分钟内达到了潜在治疗相关左旋多巴血浆浓度;·与口服施用和对于肺部施用所预期的相比,吸入后受试者与受试者之间血浆左旋多巴浓度的变化性相当低;·全身性左旋多巴暴露与施用的左旋多巴细颗粒剂量成比例;·药代动力学建模表明与口服施用相比吸入的90/8/2具有更短的滞后时间和更快的吸收速率;·与口服施用相比,吸入后剂量归一化的(基于估计的细颗粒剂量)暴露基于AUC大1.3至1.6倍,基于Cmax大1.6至2.9倍;·在没有卡比多巴预治疗的情况下,血浆左旋多巴清除率高约4倍,并且左旋多巴暴露降低。引言在本实施例中,将90/8/2作为患有帕金森病的患者中运动波动(“off发作”)的发作治疗进行测试,所述患者对于其标准口服药物具有间歇性不充分响应。90/8/2可用作患者现有包含多巴脱羧酶抑制剂(即,卡比多巴或苄丝肼)的帕金森病药物方案的辅助。本研究是利用90/8/2在人中进行的首次研究,并且被设计来在成年健康志愿者中评估与口服左旋多巴相比施用90/8/2后的安全性、耐受性和左旋多巴药代动力学(PK)。已经在临床试验中测试了安全性和耐受性结果。本PK数据分析提供了对单吸入剂量的90/8/2之后左旋多巴PK的描述以及与禁食或进食条件下口服施用左旋多巴(LD;L-多巴)的比较,以及具有和不具有卡比多巴(CD)预治疗的情况下左旋多巴PK的比较。如组合的卡比多巴/左旋多巴制剂中常规描述的那样施用口服左旋多巴。研究设计和目的这是如下在健康成年男性和女性受试者中进行的二部分研究:·部分A:与口服左旋多巴相比的剂量递增方案。·部分B:90/8/2±卡比多巴预治疗方案本研究A部分的主要药代动力学目的是在健康成年人中研究施用单吸入剂量的90/8/2后左旋多巴的药代动力学。次要目的是探究单吸入剂量施用后左旋多巴的剂量成比例性(doseproportionality)并且将90/8/2的PK与禁食状态或进食状态施用的口服左旋多巴相比较。部分B的目的是比较具有和不具有卡比多巴预治疗的情况下90/8/2的耐受性和药代动力学。部分A是开放标记、3阶段交叉、单递增剂量研究。在研究药物治疗的前一天和当天,用口服卡比多巴治疗所有受试者。每位受试者在一个时期在进食或禁食状态下接受单口服剂量的CD/LD(25/100mg),在两个不同时期接受单递增剂量的两种不同吸入剂量的90/8/2。登记两组,每组9位受试者。部分A的研究设计概括在下表1中:表1:部分A研究设计。*用于90/8/2施用的左旋多巴剂量指示估计的颗粒剂量(FPD;即‘肺递送剂量’);口服CD/LD(25mg/100mg)。部分B是开放标记、随机化、二阶段、阶段平衡的交叉研究。在部分A的安全性和PK数据的预审之后,在具有和不具有CD预治疗的情况下,以随机、平衡方式对8位受试者评估单吸入90/8/2剂量(40mg左旋多巴FPD)施用后的安全性、耐受性和左旋多巴PK,使得相等数目的受试者接受下文定义的两种给药顺序A->B或B->A:方案A:90/8/2具有CD预治疗方案B:90/8/2不具有CD预治疗研究的部分A和部分B中的卡比多巴治疗根据表2中的方案归一化。在部分A中,在给药前和口服CD/LD施用后10min、20min、30min、45min、60min、75min、90min、120min、4h、8h、16h和24h采集血样。在部分A和部分B的90/8/2吸入治疗时期期间,在相同时间采集样品并且加上1、2和5分钟的额外样品。使用经验证的液相色谱-串联质谱(LC-MS-MS)测定通过SimbecResearchLimited确定血浆左旋多巴浓度,定量的下限为9.84ng/mL(2,3)。表2:卡比多巴治疗方案。*当口服和吸入给药时期按计划在两个连续日发生时,第一给药时期执行的CD给药方案充分包括了第二给药时期所需的CD预治疗。在接受研究药物前一天期间,部分A和部分B(+CD)中的受试者接受3个剂量的CD。**不适用于随机化进食状态的受试者。***注意:25mg卡比多巴也在T0施用,作为口服CD/LD施用的一部分。药代动力学分析方法非区室分析对每位受试者和每种治疗的血浆浓度和时间进行数据分析,利用专业版本5.3进行非区室分析。使用线性梯形法(lineartrapezoidmethod)评估从时间零到最后可测量时间点的曲线下面积(AUC0-t)。最后三个或更多个时间点的线性回归用于评估消除速率常数(λ),后者用于由以下方程式评估终末半衰期(T1/2)和从0到无穷的AUC(AUC0-∞):T1/2=ln(2)/λAUC0-∞=AUC0-t+Ct/λ其中Ct是通过回归线预测的最后可测量浓度。由以下方程式评估血清清除率除以生物利用度(CL/F)和终末阶段的表观分布体积除以生物利用度(Vz/F):CL/F=剂量/AUC0-∞Vz/F=剂量/(λ*AUC0-∞)直接由数据确定最大浓度(Cmax)和观察到它的最大时间(Tmax)。通过梯形法计算药物施用后最初10分钟的部分AUC(AUC0-10m)。确定最初10分钟观察到的最大血浆浓度(Cmax,10m),作为从给药到包含10分钟取样时间所观察到的最高血浆浓度。通过用90/8/2吸入后的剂量归一化Cmax或AUC除以口服施用后的剂量归一化参数来计算每个受试者的吸入与口服暴露比。基于AUC的暴露比是吸入药物对口服药物的相对生物利用度。利用包括由Cmax除以2计算的血浆浓度的两个血浆浓度的时间点之间的线性插值计算(MicrosoftExcel)额外参数——达到观察到的最大血浆浓度的一半的时间(TCmax50)。药代动力学建模使用专业版本5.3进行药代动力学建模。评估若干不同模型,包括具有和不具有滞后时间的一和二区室模型。所有评估模型具有一级输入。基于若干诊断标准评估模型,包括Aikaike信息标准,残差平方和,估计参数及其各自估计标准误差的相对值,观察浓度和预测浓度的相关性,以及预测浓度和观察浓度之间变化的一般趋势。最佳描述了大部分血浆浓度对时间曲线的模型是具有滞后时间的二区室模型(模型12)。还通过不具有滞后时间的模型描述了来自接受吸入90/8/2的受试者的大部分数据组,这是因为从这些受试者估计的滞后时间非常短,在大部分情况下小于1分钟。但是,为了与来自口服施用的数据组进行比较,滞后时间模型用于所有受试者和所有治疗。与一区室模型相比,二区室模型更好地描述了大部分数据组。在一些情况下,一区室模型可能不合适。在一区室模型更好的情况下,基于统计学诊断标准,两个模型之间的差异非常小。因此,本文给出了使用二区室模型建模的结果。二区室模型方案1的模型产生以下的估计:分布体积除以吸收的剂量分数(V/F)、滞后时间(Tlag)和与吸收和消除有关的速率常数(分别为k01和k10),以及区室间速率常数k12和k21。由k12、k21和k10计算与曲线的吸收和消除阶段有关的速率常数α和β。由一级参数计算的其他二级参数包括AUC、Cmax、Tmax、CL/F以及与曲线的吸收、分布和消除阶段有关的半衰期(T1/2k01、T1/2α、T1/2β)。模型由以下方程式表示:Ct=Ae-αt+Be-βt+Ce-k01tCt是施用后时间t的血浆左多巴浓度,A、B和C是曲线的分布、消除和吸收阶段的y轴截距并且由剂量、体积和速率常数计算。方案1在所有分析中使用均匀加权(Uniformweighting),并且报告为低于测定的定量水平(BLQ,<9.84ng/mL)的血浆浓度作为缺失值处理。分析中未排除数据点。结果和讨论通过吸入以10至50mg左旋多巴FPD的剂量施用的90/8/2产生了快速升高的与剂量成比例的血浆左旋多巴浓度,在健康成年人中20至50mg左旋多巴的细颗粒剂量后5至10分钟内实现了潜在的治疗相关水平(400至500ng/mL)。图1表示90/8/2吸入之后以及在进食和禁食条件下的100mg口服剂量之后的平均左旋多巴血浆左旋多巴浓度。对于分别为10mg、20mg、30mg和50mg左旋多巴的每个吸入剂量以及在进食和禁食条件下并且在具有和不具有卡比多巴预治疗的情况下口服100mg左旋多巴,计算个别数值和浓度对时间曲线。90/8/2吸入后的血浆左旋多巴浓度比禁食条件下口服施用后的那些更快增加,比进食条件下的那些快得多。在90/8/2吸入后约5分钟,达到了潜在的治疗相关血浆浓度。在吸入20至50mgFPD的90/8/2后的5分钟内,血浆浓度为400至500ng/mL或更高,观察的潜在治疗相关的范围(4)。在40和50mgFPD的90/8/2之后达到的血浆浓度与在口服CD/LD(25/100mg)给药后观察到的那些在相同范围(图3).图2示出了与口服施用后的那些相比最初10分钟的平均血浆浓度。药物施用后最初10分钟的暴露表示为表3中0至10分钟的AUC(AUC0-10m)和在最初10分钟观察到的最大血浆浓度(Cmax,10m)两者。在一些个体中,在不到10分钟观察到Cmax,10m。禁食状态下口服施用导致与进食状态下相比更快速的吸收,但是依然比吸入之后慢得多。如文献(5)中所述,在口服施用之后,在进食受试者中观察到了相当大的Cmax降低和Tmax延长,但是,进食和禁食受试者之间AUC(表5)类似。表3:90/8/2吸入或口服左旋多巴施用后的左旋多巴暴露与口服施用之后相比,90/8/2吸入之后的治疗后受试者间血浆浓度变化性低得多。参见图3,在吸入之后(实心符号),大部分接受50mg90/8/2的受试者中的血浆浓度在给药后10分钟高于400ng/mL,一些在5分钟且全部到20分钟高于400ng/mL。在口服施用之后(空心符号),响应低得多,没有受试者在给药的10分钟内接近400ng/mL。其他剂量组的个体血浆浓度和变化性数据表明,在20mg和以上的左旋多巴FPD剂量下,在一些受试者中在给药的5至10分钟之内达到高于400ng/mL的血浆浓度,并且响应变化性比口服施用之后低得多。表4中示出的给定取样时间治疗组内表示为%CV的血浆浓度变化性的程度证明,在给药的最初30分钟内,90/8/2治疗受试者中的变化性低于禁食口服组中观察值的一半,为全口服受试者(进食和禁食组合)中的约五分之一。表4:血浆左旋多巴浓度的变化性(%CV)*是指估计的左旋多巴细颗粒剂量**口服左旋多巴剂量100mg表5中示出了通过非区室分析评估的药代动力学参数的概述。对于分别为10mg、20mg、30mg和50mg的每个吸入剂量以及在禁食和进食条件下并且具有和不具有CD预治疗的情况下的100mg口服剂量,由非区室PK分析确定个体的参数估计。结果表明左旋多巴暴露与施用的90/8/2剂量成比例。对于所有90/8/2剂量,剂量归一化Cmax和AUC非常类似。图4和图5中进一步说明了剂量成比例性。所有剂量的T1/2类似。表5:通过非区室分析评估的左旋多巴药代动力学参数(平均值±SD)*剂量:左旋多巴剂量**是指估计的细颗粒剂量***中值由剂量归一化AUC0-∞的比计算个体受试者的吸入90/8/2相对于口服左旋多巴的生物利用度。由于研究的部分A中的每位受试者接受一个口服剂量和两个吸入剂量,所以确定每位受试者的两个生物利用度估计值,每个吸入剂量一个。还在剂量归一化Cmax值上进行相对暴露计算。对于在进食和禁食条件下施用的口服剂量,分开进行计算。表6中示出了用于相对生物利用度计算的平均值和标准偏差。计算个别值,作为由剂量归一化Cmax计算的与卡比多巴/左旋多巴25/100mg)口服施用相比吸入90/8/2(10-50mg左旋多巴细颗粒剂量)之后的相对左旋多巴暴露。似乎没有进食受试者和禁食受试者之间或剂量组之间的主要差异。与口服施用相比,吸入之后的剂量归一化的(基于估计的细颗粒剂量)暴露基于AUC大约大1.3至1.6倍,基于Cmax大1.6至2.9倍。表6:吸入90/8/2相比于口服左旋多巴的暴露比(平均值±SD)通过具有一级输入和滞后时间的二区室模型最佳描述了血浆浓度对时间曲线。在个体数据组上进行建模,并且使用模型12制作观察的和预测的浓度对时间曲线。在一些情况下,终末半衰期(T1/2β)的估计值非常大,这是因为曲线最终阶段中的一些点的浓度类似或波动,导致平缓坡度。在这些情况中的许多中,大的T1/2β产生了非常大的AUC估计值。来自模型的参数估计值中的其他变化性造成一些参数估计值中的一些异常值。这些值不从数据分析中排除或者在统计学上作为极端值处理。相反,通过中值而非平均值概况数据。因此,异常高或低的值保留在展示的数据中,但是不会不当地影响组的概况统计。表7中示出的药代动力学建模结果表明,口服施用后有约9分钟的滞后时间。相比之下,与吸入90/8/2相关的滞后时间可忽略,其小于0.5分钟。另外,吸入90/8/2的吸收速率比禁食状态下口服施用后的更快(更短T1/2k01),并且是进食状态下吸收的快约10倍。与口服施用相比,90/8/2吸入后更短的滞后时间和更快的吸收速率解释了给药后最初5至10分钟内观察到的更大的全身性暴露。计算的参数—达到50%Cmax的时间(TCmax50)还表明,90/8/2吸入产生了比口服施用更早的左旋多巴全身性暴露。除了进食状态的口服施用之外,吸收比消除快得多。图6说明了滞后时间和吸收速率对施用后最初数分钟内血浆浓度的联合效应,其示出了平均血浆浓度数据的药代动力学建模。该图示出了给药后最初60分钟90/8/2吸入和口服左旋多巴施用的通过药代动力学模型预测的浓度。符号表示观察到的平均浓度,线表示通过药代动力学模型预测的浓度。预测值和观察值的良好相关性表明模型非常好地描述了数据。图还说明了来自研究的其他观察结果,90/8/2吸入导致血浆左旋多巴浓度快速升高,在给药的5至10分钟内可以达到潜在临床相关血浆浓度,并且暴露与剂量成比例。表7:通过药代动力学建模评估的药代动力学参数(中值)*指估计的细颗粒剂量部分B图7示出了来自研究的部分B的血浆浓度,其中在交叉设计中具有或不具有卡比多巴预治疗的情况下吸入40mg左旋多巴FPD的90/8/2。利用卡比多巴预治疗时峰值血浆浓度和暴露更高。血浆左旋多巴清除比没有CD预治疗时快约4倍。相应地,在没有CD预治疗的情况下,Cmax和AUC更低,Tmax和T1/2稍微更短(表8)。表8:在具有和不具有卡比多巴预治疗的情况下吸入40mg90/8/2后通过非区室分析评估的左旋多巴药代动力学参数(平均值±SD)*中值结论本研究的主要发现是:(i)吸入90/8/2导致血浆左旋多巴浓度快速升高;(ii)与口服药物施用相比,利用90/8/2吸入给药后最初10分钟基于Cmax和AUC的全身性左旋多巴暴露高得多;(iii)在健康成人中,20至50mg左旋多巴细颗粒剂量的90/8/2给药后的5至10分钟内达到了潜在治疗相关血浆左旋多巴浓度;(iv)与口服施用相比,吸入后受试者与受试者之间血浆左旋多巴浓度的变化性相当低;(v)全身性左旋多巴暴露与施用的左旋多巴细颗粒剂量成比例;(vi)药代动力学建模表明与口服施用相比吸入的90/8/2具有更短的滞后时间和更快的吸收速率;vii)与口服施用相比,吸入后剂量归一化的(基于估计的细颗粒剂量)暴露基于AUC大1.3至1.6倍,基于Cmax大1.6至2.9倍;以及viii)在没有卡比多巴预治疗的情况下,血浆左旋多巴清除率高约4倍,并且左旋多巴暴露降低。实施例2测试两个剂量的肺部左旋多巴(25mg和50mg研究药物)的阶段2研究是多中心、随机化、双盲、安慰剂对照、单剂量、交叉设计,其具有三个臂(安慰剂、25mg和50mg)并且包括“开放标记”的口服Sinemet臂。对本研究中治疗的24位PD(24)患者在每次访问时的L-多巴血浆水平、运动响应(motorresponse)和安全性进行连续评估。在OFF状态向患者施用研究药物,连续评估在给药前开始并且持续至给药后180分钟。使用敲击测试、帕金森病统一评定量表第III部分(UPDRSIII)和主观评价“有意义的”NO和OFF来测量运动功能。监测的安全性参数包括肺功能、临床实验室数据、EGC和生命体征(血压、心率和直立血压(orthostaticbloodpressure))。本研究设计为测量肺部左旋多巴对运动功能的影响的时间、大小和持久性,以评估帕金森病患者中肺部左旋多巴的安全性和耐受性。在药代动力学参数和药效学参数的比较中,本发明人发现了处于off状态的患者和处于on状态的患者之间出人意料的陡曲线。在图8中,将患者的血浆左旋多巴浓度与UPDRS评分进行了比较。UPDRS是用于帕金森病患者的标准测试,以测试其对于药物治疗的响应及其疾病进展。由图8可以看到,处于on的患者和处于off的患者之间存在非常小的左旋多巴血浆浓度差异。低至200-400ng/ml的左旋多巴血浆浓度造成了off状态和on状态之间的差异。真正显著的是四位不同患者在这里所表现的,其全部具有显著不同的基线左旋多巴血浆浓度。不同的基线左旋多巴血浆水平涉及每位患者具有不同的有效剂量或有效浓度以使得左旋多巴对于每位患者具有效果的事实。尽管患者群体之间具有不同有效剂量或有效浓度,从off到on所需的血浆浓度的升高非常小。实施例3阶段2(b)随机化、双盲安慰剂对照的研究利用90/8/2的阶段2b研究设计和方法本研究是用于经历运动波动(OFF发作)的帕金森病(PD)受试者中每日多至3次OFF发作的治疗的吸入(吸入的左旋多巴[LD]粉末)或安慰剂的随机化、双盲、安慰剂对照的多中心研究。将受试者以1:1比例随机分配为接受吸入90/8/2(在本文中也称为“研究药物”)或安慰剂;通过受试者的Hoehn和Yahr阶段(<2.5对≥2.5)分级来随机化,以平衡每组中疾病的严重程度。90/8/2LDFPD包含含有90%LD、8%二棕榈酰磷脂酰胆碱(DPPC)和2%氯化钠(NaCl)的均匀颗粒。使用如美国专利No.8,496,002中所述的用于粉末吸入的吸入器装置递送90/8/2,所述专利通过引用并入本文。90/8/2提供在尺寸00的羟丙甲纤维素(羟丙基甲基纤维素[HPMC])胶囊中,各自标称填充重量为32mg(每个胶囊27.6mgLD),设计为向肺递送约可呼吸剂量的LD17mgFPD。两个选择的90/8/2剂量水平(约34mgFPD和50mgFPD)为基于健康成年志愿者中的研究的安全性和药代动力学数据(PK)数据和本文实施例1的安全性,来自实施例1中在健康成年志愿者中进行的研究的PK,以及来自如实施例2中所述在PD患者中进行的研究的安全性、PK和药代动力学数据。为了保持盲性,给予所有患者看起来相同的研究药物试剂盒,并且指导每剂量吸入相同数目的胶囊(第1周和第2周期间2个胶囊,第3周和第4周期间3个胶囊)。如需要,允许患者在胶囊吸入之间啜饮水。安慰剂吸入粉末提供在尺寸00的HPMC胶囊中,各自为10mg的标称填充重量。安慰剂吸入粉末为吸入级乳糖一水合物NF。选择乳糖的颗粒大小以提供可比较的吸入粉末的头部沉淀和模拟吸入的感觉。研究中检查两个90/8/2剂量水平:剂量水平1(DL1)为每个治疗时期约35mgLD细颗粒剂量(FPD),剂量水平2(DL2)为约50mgLDFPD。盲性吸入研究药物DL1的第一剂量在访问3时在诊所给予(即,吸入90/8/2或安慰剂的2个胶囊);每个90/8/2胶囊递送约17.5mgLDFPD。盲性研究药物DL2的第一剂量在访问5时在诊所给予(即,吸入90/8/2或安慰剂的3个胶囊)研究具有3个时期:筛选、治疗和随访,总共7次访问(2次筛选访问、4次治疗访问和1次随访访问)。对于每位受试者,治疗期为约4周,研究持续时间为约8周至10周。每位患者每天自己施用吸入研究药物的多至3个剂量,持续4周。从筛选直到最终研究访问,不允许受试者的通常PD药物的剂量或给药方案变化。受试者资格年龄30至80岁的男性和女性受试者如果符合以下条件则有资格参与本研究:在30岁后具有诊断的特发性PD;满足UKBrainBank标准的步骤1和2;在ON状态被归类为改进的Hoehn和Yahr阶段1-3;每个清醒天经历自我报告的和由PD日志确认的最少2小时的平均每日OFF时间(排除清晨OFF时间)的运动波动;并且表现出可接受的LD响应性。受试者在筛选访问1之前必须处于稳定的含LD口服治疗剂量/方案至少2周;含LD/多巴胺脱羧酶抑制剂(DDI)的方案必须包括在清醒天至少施用4次的给药方案。受试者在筛选访问1之前对于至少4周的其他PD药物应是稳定的。在筛选时受试者在OFF状态和ON状态之间记录的UPDRS第3部分评分之间应具有≥25%差异。受试者必须已经理解(具有或不具有护理人员帮助)并且在研究期间不改变其每日药物剂量。受试者必须具有正常认知,如通过细微精神状态检查(Mini-MentalStateExamination,MMSE)≥25的评分确定的。在筛选时的ON状态,如通过呼吸量测定法预测的,受试者必须已具有>预测的60%的筛选FEV1和≥预测的75%的FEV1/FVC比,且没有肺病史。评估标准和终点表9中描述了研究目的和变量。表9研究目的和变量缩写:C-SSRS=Columbia自杀严重性评级量表;PD=帕金森病;PDQ-39=39-项目帕金森病问卷;PGI-C=患者变化的整体印象(PatientGlobalImpressionofChange);QUIP=帕金森病中冲动/强迫性障碍问卷;UPDRS=帕金森病统一评定量表。效力由诊所和在家(门诊)评估二者评估,如通过以下标准概况的:诊所标准:UPDRS第3部分运动评分;在诊所施用研究药物后OFF发作消退至ON状态的时间(每次检查员评估);研究药物施用后运动障碍的出现、持续时间和严重程度。在家标准:受试者报告的研究药物施用后OFF发作消退至ON状态的时间(来自吸入药物治疗记录表),每日无运动障碍ON时间、具有运动障碍(麻烦的和无麻烦的)ON时间和OFF时间的PD日志信息。进行以下探究性评估:PGI-C、PDQ-39、如上所述效力标准(评估90/8/2治疗臂中2个剂量水平之间的潜在差异)。安全性由体检、不良事件(AE)报告、标准和直立性生命体征(血压和心率)、呼吸率、临床实验室值(血液学、生物化学和)、心电图(ECG)以及用于评估肺功能的呼吸量测定法。此外,用于评价自杀倾向、嗜睡和冲动控制行为的评估在基线和随访时进行。基线特征八十六位患者(86)参与本研究。在访问2之前的3个连续日中,患者完成筛选PD日志,记录其清醒ON/OFF状态(时间OFF、没有运动障碍的时间ON、具有无麻烦运动障碍的时间ON、具有麻烦运动障碍的时间ON)和睡眠时间。此外,患者将在访问2之前的7天完成筛选ON/OFF发作和药物记录表,记录清醒日经历每次OFF发作的以下信息:OFF发作开始的时间、下一次ON开始的时间以及他们是如何使用他们的标准LD药物。尽管患者约每三个小时进行其当前左旋多巴治疗方案,这些患者报告OFF为其清醒时间的约1/3至1/2(参见表10和11)。表10全部(n=86)年龄(岁)62.4(8.7)自PD的时间Dx(年)9.4(3.9)LD的持续时间Tx(年)7.8(3.9)自波动出现的时间(年)4.3(3.6)每天平均OFF时间(小时)3.5(1.5)–自我5.7(1.8)-PDD*每天平均#OFF发作3.6(1.1)*~1h清晨OFF(总每日OFF时间的15-20%)表11因此,很明显OFF时期的管理依然是PD患者中重要的未满足的需求,如表12和13中指出的。表12提供了在接受研究药物或安慰剂之前每位患者的平均给药方案。表12表13提供了平均每日OFF和ON时间,如在接受研究药物或安慰剂之前在日志中由患者报告的。表13*PD日志中至少2天的任何运动障碍≥1hr/天在阶段2b研究的筛选阶段和任一组接受研究药物或安慰剂之前测量基线UPDRS第3部分评分。数据示于表14。表14暴露于研究药物或安慰剂的记录的总研究天数为约6.5患者年数(patientyears)(2,369患者天数)。经治疗OFF期的总数等于施用的总剂量,为约4,484,安慰剂施用2314剂量,研究药物施用2369剂量。用于整个研究的安慰剂或研究药物的总胶囊为11,115。一位患者由于麻烦的运动障碍而经历了剂量降低,但是该患者施用的是安慰剂。由于恶心,给予研究药物中的一位患者剂量降低。基于患者自己的记录,每天研究药物的平均使用为每天使用2.1次,其排除早晨“OFF”时间。结果阶段2b研究的主要终点是在访问6(DL2)评估与给药前相比UPDRS第3部分评分的平均变化(给药后10-60分钟)中研究药物对安慰剂的差异。使用DL1的访问4和访问5(DL2的第一剂量)中的相同差异用作次级终点。与UPDRS一致,UPDRS运动评分的临床重要差异(CID)是最小为2.5个点,中等为5.2个点,大CID为10.8个点(Shulman等,ArchNeurol,第67卷(2010年1月))。90/8/2满足在50mg剂量访问6时与安慰剂相比平均UPDRS第3部分运动评分(给药后10-60分钟的时期)的统计学上显著降低的主要终点。在60分钟时间点,87%的患者实现了UPDRSIII的临床有意义的降低。此外,在访问4、5和6评估两种测试剂量(35mg和50mg)的所有时间点(即,10、20、30和60分钟),90/8/2显示出临床相关的和统计学显著的降低。在早至给药后10分钟,临床相关UPDRSIII改善就已经明显。在访问6,用90/8/2(50mg剂量)治疗之后,维持临床显著响应直至给药后至少60分钟。在每个给药后时间点,90/8/2和安慰剂之间的差异是统计学显著的。表15提供了初筛以及访问4(V4)、访问5(v5)和访问6(v6)时研究药物组和安慰剂组之间给药前UPDRS评分的比较。在访问4时递送剂量水平1(DL1),而在访问5和访问6时递送剂量水平2(DL2)。表15如图9和10中所示,在每个时间点,以50mgFPD施用的研究药物(图9)和以35mg施用的研究药物(图10)之间具有统计学差异。表16提供了访问4-6各自的平均最佳UPDRS第3部分变化。平均值(SEM)*所有访问统计学显著差异表17示出了访问4-6各自的平均最佳百分比(%)UPDRS第3部分变化。平均值(SEM)*所有访问统计学显著差异总结根据本文提供的数据,阶段2b研究达到了其主要终点,表现为与安慰剂相比在访问6的给药后10至60分钟平均UPDRS第3部分评分与给药前相比具有统计学显著的平均变化。数据还表明UPDRS时间曲线的形状指示快速和持久的响应,35mg剂量(DL1)具有与50mg剂量(DL2)类似的振幅,但是效果的持续潜在地稍微更短。所有访问和剂量中UDPRS第3部分评分的最佳变化和最佳百分比变化也是统计学显著的,甚至在安慰剂响应衰减之前的V4(DL1),具有显著分离。数据一起表现出每日OFF时间的强烈的临床有意义和统计学显著的改善,而不增加具有运动障碍的ON时间,如图11所示。图11示出了报告与安慰剂组相比在研究药物组中测试的患者的麻烦的和无麻烦的运动障碍的数据(如在其各自PD日志中自我报告的)。实施例1和2以及本实施例的研究还表明,在所有阶段1、2a和2b研究测试的所有剂量水平,药物是安全的并且良好耐受。本文引用的所有专利和科技文献建立了本领域技术人员可获得的知识。本文引用的所有美国专利和公开的或未公开的美国专利申请通过引用并入本文。本文引用的所有公开的外国专利和专利申请通过引用并入本文。所有其他公开的文献、文档、手稿和科技文献通过引用并入本文。尽管参考本发明的优选实施方案特别示出和描述了本发明,本领域技术人员将理解的是,可以在其中进行各种形式和细节的变化,而不脱离由所附权利要求包括的本发明的范围。还应该理解,根据本发明,本文描述的实施方案不互相排斥,并且来自多个实施方案的特征可全部或部分组合。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1