治疗骨肉瘤的雷公藤甲素衍生物及其制备方法与流程
本发明属于医药领域,具体地,本发明涉及一种用于治疗骨肉瘤的雷公藤甲素衍生物及其制备方法。
背景技术:
骨肉瘤是骨恶性肿瘤中最多见的一种,是从间质细胞系发展而来,肿瘤迅速生长是由于肿瘤经软骨阶段直接或间接形成肿瘤骨样组织和骨组织。骨肉瘤病理确诊后需要即刻进行放化治疗,截肢手术则作为传统切除肿瘤组织的重要方法。该方式对病情起到了较好的控制效果,但对患者的身心伤害较大,严重影响术后生存质量。虽然通过新辅助化疗结合手术治疗,其预后有较大改善,5年生存率由20世纪70年代末的10%左右上升到如今的60%左右,但近30多年间,骨肉瘤经手术治疗3年后有30-40%的复发率,尽管进行了多种努力,一直未能取得较大进展。
目前临床常规使用的化疗主要是以大剂量甲氨喋呤、顺铂、阿霉素、异环磷酰胺为主。化学治疗改善了部分骨肉瘤的治疗效果,减少了术后痛苦,提高了术后生存率。
雷公藤甲素(Tripodile,TP)是从传统中药雷公藤中分离得到的环氧化二萜内酯化合物,又名雷甲素,是雷公藤的主要有效成分之一。雷公藤甲素具有极其显著的抗肿瘤活性。研究表明,雷公藤甲素是一种广谱肿瘤抑制剂,体外可诱导多种肿瘤细胞凋亡,包括卵巢癌、乳腺癌、结肠癌、口腔癌、胃癌等,体内可抑制肿瘤生长和肿瘤细胞转移,包括血液肿瘤、恶性肿瘤和实体瘤。雷公藤甲素抗肿瘤活性优于顺铂、阿霉素、紫杉醇等传 统抗癌药物,在极低浓度(2-10ng/ml)下即可有效抑制肿瘤细胞增殖。除此之外,雷公藤甲素还可避免肿瘤耐药,同时提高肿瘤细胞对其它抗癌药物的敏感性,与化疗药物和电离辐射相结合发挥协同效应。
凌昌全等经研究发现雷公藤甲素能够显著抑制多种骨肉瘤细胞的增殖,并且能够诱导其发生凋亡;激活线粒体相关信号通路;显著抑制骨肉瘤在裸鼠体内生长,并检测到细胞凋亡现象[石倩玮,李曙光,李俊,凌昌全.雷公藤甲素抗骨肉瘤细胞的实验研究.2013,33,659-663]。
尽管雷公藤甲素具有显著的抗骨肉瘤活性,但其本身水溶性差,生物利用度低,毒性较强,治疗窗窄。同时雷公藤甲素对肿瘤细胞和正常细胞没有选择性,带来了很大的毒副作用。因此如何提高其水溶性,增强其靶向性研究就成为一种必然。
任天斌等在聚乙二醇的两端通过二硫键连接上雷公藤甲素,该偶合物在一般的体液环境中是稳定存在的,雷公藤甲素由于疏水性构成疏水内核,外壳为亲水性的聚乙二醇,一旦达到病变位置,由于还原环境的刺激,二硫键开始断裂,从而快速释放出内核中的雷公藤甲素[CN10243866]。
Georg等人在雷公藤甲素14-位羟基上上引入亲水性的基团增强其水溶性[WO2010129918]。Dai等人也提出了一些溶解度提高和毒性降低的雷公藤内酯前药[US6548537]。
然而,关于靶向性雷公藤甲素前药的研究报道较少,且这些增强雷公藤甲素水溶性的前药也存在着许多缺陷。比如雷公藤内酯琥珀酸酯前药的不完全转化[European Journal of Cancer 2009,45,1764-1772]等等。
因此,开发一种具有靶向性的、同时能增强雷公藤甲素水溶性的前药势在必行。
技术实现要素:
针对以上技术情况,本发明提供一种新的雷公藤甲素衍生物,所述衍生物为如下式(I)的偶合物:
其中,R1=为H或OH;R2=为H或OH;R3=为H或OH;R4=为H或OH;R5=为H或OH;R6=为H或OH;R7=为H或OH;G为O或NH;M为O或OH;X为O或OH;
取代基S1为对氨基芳甲氧羰基、对氨基芳甲氧基,N,N’-二甲基-N-羰基乙二胺、对羟基芳甲氧羰基、对羟基芳甲氧基、对巯基芳甲氧羰基、对巯基芳甲氧基、取代巯基丁酰基、取代巯基戊酰基、巯基乙氧羰基或巯基丙氧羰基;
取代基L为硫硫连接键、多肽连接键或糖苷连接键;
取代基S2为琥珀酰亚胺烷基酰基、烷基二酰基,或者琥珀酰亚胺烷基氨基二酰基;
A为适配子;
其中S1与M或X相连;其中当S1与M相连时、M为O、X为O、X与C18间为双键且C3-C4间为双键,或者当S1与M相连时、M为O、X为OH、X与C18间为单键,且C18-C3间及C19-C4间为双键;其中当S1与X相连时,M为OH、X为O、X与C18间为单键、且C18-C3间及C19-C4间为双键。
本领域技术人员由雷公藤甲素的化学结构不难知道,该结构存在互变异构体,因此基于雷公藤甲素及其类似物的已知结构互变的特性而发生在 本发明式(I)偶合物中的结构互变,这都属于本领域技术人员基于本领域常识及雷公藤甲素已知特性所能预期出的;除非在本发明式(I)偶合物结构中失去了互变异构的条件;作为示例性的说明,即本发明式(I)中S1与M相连时、M为O、X为O、X与C18间为双键且C3-C4间为双键的化学结构与当S1与M相连时、M为O、X为OH、X与C18间为单键,且C18-C3间及C19-C4间为双键的化学结构实际为互换结构,属于互换异构体,其中主要是以M为O、X为O、X与C18间为双键且C3-C4间的化学结构存在;而当S1与X连接时,X为O,此时该部位失去了互变异构的条件;但本发明式(I)衍生物的其他具有结构互换特性的部位由于其结构特性而产生了互变异构体,其同样是基于雷公藤甲素已知的结构互换的特性,这也是本领域技术人员结合本领域常识所能预期出的,均没有超出本发明所保护的范围。
作为本发明实施方案之一,所述取代基L为硫连接连接键时,所述硫硫连接键结构如下:-S-S-。
作为本发明实施方案之一,所述取代基L为多肽链接键时,所述多肽连接键为二至四个氨基酸连接的多肽;
作为本发明实施方案之一,所述多肽链优选为缬氨酸和瓜氨酸,苯丙氨酸和赖氨酸,丙氨酸和赖氨酸,缬氨酸和赖氨酸,苯丙氨酸和精氨酸,苯丙氨酸和瓜氨酸,亮氨酸和瓜氨酸,异亮氨酸和瓜氨酸,或色氨酸和亮氨酸形成的二肽。
作为本发明实施方案之一,所述L为所述的糖苷连接键时,所述的糖苷连接键为β-葡醛酸。
作为本发明实施方案之一,所述的S2具有如下结构:琥珀酰亚胺烷基酰基(式i)、烷基二酰基(式ii)、或者琥珀酰亚胺烷基氨基二酰基(式iii):
n为1-10的正整数,优先选择4-6的正整数,例如n为1、2、3、4、5、 6、7、8、9或10的整数,m为1-10的正整数,优先选择4-6的正整数,例如m为1、2、3、4、5、6、7、8、9或10的整数,式(i)~式(iii)中的A为本发明中的适配子,L为本发明中的连接键。
作为本发明实施方案之一,所述的适配子A可以是为本领域能够靶向骨肉瘤细胞的任何的适配子;
作为本发明实施方案之一,所述适配子A包括但不限于具有SEQ ID NO.1、SEQ ID NO.2或SEQ ID NO.3所示的核苷酸序列,其中
SEQ ID NO.1:
GTACTTCCGACTTGGGGCGGGTTTGTTGCTGGCTGTCGGCAAAAGTGCA,简称“LC4”;
SEQ ID NO.2:
GTACTTCCGGCGGGTTCTATGGGCCCTGTCTCCCTTCCCAAAAGTGCACGCTAC,简称“LC5”;
SEQ ID NO.3:
GTACTTCCCGCGTGTGTGTGTTTGTCGGAGTGTTATCGCAAAAGTGCACGCTAC,简称“LC6”。
作为本发明实施方案之一,上述适配子可采用本领域常规的制备方法进行制备,本发明包括但不限于以下方法:
1)适配子的筛选
准备上述随机ssDNA文库、FITC标记的正向引物和Biotin标记的反向引物;
选择大鼠成骨细胞为靶细胞,大鼠肝细胞系和外周血单核细胞为非靶细胞,采用无酶的细胞消化液处理生长良好的细胞,接种于培养皿中,过夜培养,当细胞长至95%时即可用于文库筛选;
将靶细胞消化,并与随机文库用孵育缓冲液孵育;
孵育结束后将细胞离心下来,加入双蒸水,收集上清,上清中含有第一轮筛选与靶细胞结合的单链DNA,用于PCR扩增,制备下一轮筛选亚文库;
PCR产物dsDNA经浓缩管浓缩后,与链亲合素-磁珠进行孵育,加入 碱性溶液进行碱变性,使得Biotin-ssDNA与链亲合素结合后固定在磁珠上,游离的FITC-ssDNA亚文库则被分离出来;然后取上清,加入酸溶液进行中和;
将上述FITC-ssDNA亚文库进行高分辨率的琼脂糖电泳,再次分离FITC-ssDNA及其中的污染序列(dsDNA);
溴化乙锭染色10分钟,紫外灯下切取FITC-ssDNA的目标条带,通过小分子量DNA纯化回收试剂盒进行回收;
2)适配子的富集
仅在第一轮筛选中对靶细胞进行孵育,从第二轮开始,正筛以后引入非靶细胞负筛过程;最后离心收集上清,弃去与非靶细胞结合的序列,重复上述过程5-20个循环;
3)适配子的测序:
将最后富集到的文库PCR扩增成双链DNA,使用克隆试剂盒进行克隆,经蓝白斑筛选,挑选克隆进行测序;根据测序结果使用RNA structure软件和DNAMAN软件进行一级序列和二级结构的分析,即得。
作为本发明实施方案之一,所述方法步骤1)中的孵育缓冲液可以本领域常用的适于本发明的各种缓冲溶液,本发明优选为含有1.0mM MgCl2,0.1mg/ml酵母tRNA和0.1mg/ml鲑鱼精DNA的磷酸缓冲溶液;
作为本发明实施方案之一,所述方法步骤1)中PCR的反应条件可以有本领域技术人员结合本发明和本领域技术人员进行确定,作为优选实施方案之一,本发明优选PCR反应条件为:94度预变性3min,94度变性30s,56度退火30s,72度延伸30s,最后72度延伸5min。
作为本发明实施方案之一,所述方法步骤1)中的碱溶液可以选用本领域常用的碱性溶液,本发明包括但不限于碱溶液为200mM的NaOH溶液;
作为本发明实施方案之一,所述方法步骤1)中的酸溶液可以选用本领域常用的酸性溶液,本发明包括但不限于酸溶液为100mM盐酸。
作为本发明实施方案之一,所述方法步骤1)中的电泳条件为:琼脂糖电泳,电压设定为180V,电泳时间为40分钟。
作为本发明实施方案之一,所述特异性靶向骨肉瘤细胞的适配子的制 备方法进一步包括:
1)适配子的筛选
准备上述随机ssDNA文库、FITC标记的正向引物和Biotin标记的反向引物;
选择大鼠成骨细胞ROS17/2.8(ATCC)为靶细胞,大鼠肝细胞系BRL-3A(ATCC)和外周血单核细胞为非靶细胞,采用无酶的细胞消化液处理生长良好的细胞,按照5×106接种于培养皿中,过夜培养,当细胞长至95%时即可用于文库筛选;
将靶细胞消化下来,与随机文库在37度条件下孵育1小时,孵育缓冲液为含有1.0mM MgCl2,0.1mg/ml酵母tRNA和0.1mg/ml鲑鱼精DNA的PBS;
孵育结束后将细胞离心下来,加入双蒸水,100度水浴加热10分钟,收集上清,上清中含有第一轮筛选与靶细胞结合的单链DNA,用于PCR扩增,制备下一轮筛选亚文库;
PCR反应条件为:94度预变性3min,94度变性30s,56度退火30s,72度延伸30s,最后72度延伸5min;
PCR产物dsDNA经浓缩管浓缩后,与链亲合素-磁珠在37度孵育1小时,加入200mM NaOH溶液进行碱变性7分钟,打破dsDNA之间的氢键,使得Biotin-ssDNA与链亲合素结合后固定在磁珠上,游离的FITC-ssDNA亚文库则被分离出来;取上清,加入适量100mM盐酸溶液进行中和;
将上述FITC-ssDNA亚文库进行高分辨率的琼脂糖电泳,电压设定为180V,电泳时间为40分钟,再次分离FITC-ssDNA及其中的污染序列;
溴化乙锭染色10分钟,紫外灯下切取FITC-ssDNA的目标条带,通过小分子量DNA纯化回收试剂盒进行回收;
2)适配子的富集
为了减少ssDNA文库的损失,第一轮筛选中仅对靶细胞进行孵育;从第二轮开始,正筛以后引入非靶细胞负筛过程;正筛孵育过程与上述一致,负筛过程中,37度环境下孵育1小时以后,离心收集上清,弃去与非靶细胞结合的序列;
重复上述过程5-20个循环,为了获得较高亲和力和特异性的适配子,随着筛选轮数的增加,逐渐增加洗涤力度,洗涤次数也从3次增加到5次,洗涤时间从3分钟增加到5分钟。
筛选过程中每隔几个循环使用流式细胞仪对富集情况进行分析;将靶细胞和非靶细胞消化下来,用富集文库与其进行37度孵育1小时,PBS洗三次后,加入400ul PBS重悬细胞,流式细胞仪分析荧光强度;待荧光强度达到一定程度时,终止筛选;
3)适配子的测序:
将最后富集到的文库PCR扩增成双链DNA,使用克隆试剂盒进行克隆,经蓝白斑筛选,挑选多个克隆并进行测序,然后使用RNA structure软件和DNAMAN软件进行一级序列和二级结构的分析;根据分析结果进行截断和鉴定,即得一级序列为:
作为本发明实施方案之一,所述雷公藤甲素衍生物包括但不限于以下表中所述的化合物为:
其中,S1与M相连时、X与C18间为双键且C3-C4间为双键,或当S1与X相连时,X与C18间为单键、且C18-C3间及C19-C4间为双键。
本发明上述化合物同样会基于雷公藤甲素及其类似物的结构互变特性而发生结构互变,作为示例性的说明,即S1与M相连时、X与C18间为双键且C3-C4间为双键的化学结构可以转化为X为OH、X与C18间为单键,且C18-C3间及C19-C4间为双键的化学结构,这都属于本领域技术人员基于本领域常识及雷公藤甲素的已知特性所能预期出的;同样当S1与X连接时,此时该部位失去了互变异构的条件,但是本发明雷公藤甲素衍生物的其他具有结构互换的部位由于其结构特性而产生了互变异构体,其同样是基于雷公藤甲素已知的结构互换的特性,这也是本领域技术人员结合本领域常识及雷公藤甲素已知特性所能预期出的,其均没有超出本发明所保护的范围。
本发明所述式(I)雷公藤甲素衍生物的制备方法可由本领域技术人员根据本发明并结合本领域常识进行制备。例如包括但不限于采用如下方法制备:
一)将S1和L在极性非质子性溶剂或卤代烃溶剂中通过缩合反应或者取代反应,得到中间体一;
二)中间体一在卤代烃溶剂中和雷公藤甲素或其类似物通过不同的活化试剂缩合得到中间体二;
三)中间体二和S2在极性非质子性溶剂或卤代烃中缩合得到中间体三;
四)中间体三和适配子A在水性缓冲溶液中通过加成反应或者缩合反应进行偶联得到偶合物。
本发明方法步骤一)中,作为实施方案之一,所述极性非质子溶剂可以本领域常用的极性非质子溶剂,本发明包括但不限于二甲亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、乙醚、四氢呋喃、甲基叔丁基醚、丙酮或者它们的两种或两种以上的混合物;作为实施方案之一,所述卤代烃可以为本领域常用的卤代烃,本发明包括但不限于二氯甲烷、氯仿、四氯甲烷、二氯乙烷、四氯乙烷等或者它们两种或两种以上的混合物;
本发明方法步骤二)中,作为实施方案之一,所述卤代烃可以为本领域常用的卤代烃,本发明包括但不限于二氯甲烷、氯仿、四氯甲烷、二氯乙烷、四氯乙烷或者它们两种或两种以上的混合物;作为实施方案之一,所述活化试剂包括但不限于为芳基甲氧酰氯,如对硝基苯甲氧酰氯,苯甲氧酰氯,对甲基苯甲氧酰氯、三氯甲基苯甲氧酰氯、对氯苯甲氧酰氯、对氟苯甲氧酰氯、三氯乙腈、三氟乙腈、或DCC;
本发明方法步骤三)中,作为实施方案之一,所述极性非质子溶剂可以本领域常用的极性非质子溶剂,本发明包括但不限于N,N-二甲基乙酰胺、N-甲基吡咯烷酮、乙醚、四氢呋喃、甲基叔丁基醚、丙酮或者它们两种或两种以上的混合物;作为本发明实施方案之一,所述卤代烃可以为本领域常用的卤代烃,本发明包括但不限于二氯甲烷、氯仿、四氯甲烷、二氯乙烷、四氯乙烷等或者它们两种或两种以上的混合物。
本发明方法步骤四)中,作为实施方案之一,所述水性缓冲溶液包括但不限于磷酸盐缓冲溶液、或碳酸钠/碳酸氢钠缓冲溶液。
可选择地,本领域技术人员根据实际需要可以采用本领域常规的纯化方法,如包括但不限于反相高效液相色谱法或柱色普法对步骤四)中所得 的所述式(I)雷公藤甲素衍生物产物进行纯化,纯化条件可以由本领域技术人员结合本领域常识以及所得具体化合物的性质进行确定。
本发明以核酸适配子(Aptamer)为靶向分子,以雷公藤甲素(Triptolide)及其类似物为母体药物,采用可以在肿瘤细胞选择性断裂的硫硫键、多肽连接键以及糖苷连接键辅以不同的space连接靶向分子和母体药物,所得到的核酸适配子-雷公藤甲素偶合物具有良好的肿瘤靶向性和水溶性,因而生物利用度高,毒副作用低,抗癌活性强。本发明并通过连接键L成功地将适配子-药物偶合物链接,并使本发明耦合物不仅在循环系统中保持稳定,从而减少由于药物在循环系统中提前释放引起的毒副作用;又能使本发明耦合物在进入肿瘤细胞后,连接键断裂,能够释放出原药,从而杀死癌细胞。
实验证明,本发明以核酸适配子(Aptamer)为靶向分子,以雷公藤甲素(Triptolide)及其类似物为母体药物,采用可以在肿瘤细胞选择性断裂的硫硫键、多肽连接键以及糖苷连接键辅以不同的space连接靶向分子和母体药物,所得到的偶合物具有良好的肿瘤靶向性和水溶性,生物利用度高,毒副作用低,抗癌活性强。
附图说明
图1:不同时间点雷公藤甲素-骨肉瘤适配子偶合物在各组织中的释放率;
图2:各时间点不同组织中雷公藤甲素衍生物的浓度;
图3:肿瘤体积的测量;
图4:血清中的每次注射药物后NF-κB的mRNA表达水平。
具体实施方式
以下实施例仅用于进一步阐述本发明,但不以任何的方式限制本发明的有效范围。
适配子的制备
1.实验材料:
1.1 ssDNA文库、FITC标记的正向引物和Biotin标记的反向引物:
ssDNA文库:
5’-ATCCAGAGTGACGCAGCA-40nt-TGGACACGGTGGCTTAGT-3’
5’引物:5’-FITC-ATCCAGAGTGACGCAGCA-3’;
3’引物:5’-biotin-ACTAAGCCACCGTGTCCA-3’;均由上海生工生物工程公司提供。
1.2大鼠成骨细胞:ROS17/2.8,购自中科院细胞库,培养液为10%胎牛血清的DMEM培养基。
1.3大鼠肝细胞:BRL-3A,购自中科院细胞库,培养液为10%胎牛血清的DMEM培养基。
大鼠外周血细胞:PBMCs,直接从大鼠外周血中分离。
1.4培养液:DMEM培养基和胎牛血清购自Gbico公司
1.5流式细胞仪:购自BD Immunocytometry Systems
1.6 CH倒置光学显微镜:购自日本奥林巴斯公司
1.7 PCR试剂盒(货号:R011)、DNA Marker(20bp DNA ladder(Dye plus),货号:3420A)和琼脂糖凝胶(Agarose MS-6,货号:D615)购自大连Takara公司
1.8小分子量DNA纯化回收试剂盒(货号:ZP205)购自Beijing Zoman Biotechnology,Co.,LTD
1.9链亲合素的磁珠(货号:Z5482)购自Promega公司
1.10噻唑蓝(MTT,货号:M2128)购自Sigma公司
1.11 PCR仪购自美国Perkin Elmer公司。
1.12紫外凝胶成像系统购自Rio-Rad公司。
1.13浓缩管(Amicon Ultra-15 centrifugal Filter-10k)购自Merck Milipore公司。
1.14核酸适配子的合成由成都先导药物开发有限公司合成
2.实验方法:
2.1适配子的筛选
准备上述随机ssDNA文库、FITC标记的正向引物和Biotin标记的反向引物。
选择大鼠成骨细胞ROS17/2.8(ATCC)为靶细胞,大鼠肝细胞系BRL-3A(ATCC)和外周血单核细胞为非靶细胞,采用无酶的细胞消化液处理生长良好的细胞,按照5×106接种于培养皿中,过夜培养,当细胞长至95%时即可用于文库筛选。
将靶细胞消化下来,与随机文库在37度条件下孵育1小时,孵育缓冲液为含有1.0mM MgCl2,0.1mg/ml酵母tRNA和0.1mg/ml鲑鱼精DNA的磷酸缓冲溶液(PBS)。
孵育结束后将细胞离心下来,加入双蒸水,100度水浴加热10分钟,收集上清,上清中含有第一轮筛选与靶细胞结合的单链DNA,用于PCR扩增,制备下一轮筛选亚文库。
PCR反应条件为:94度预变性3min,94度变性30s,56度退火30s,72度延伸30s,最后72度延伸5min。
PCR产物dsDNA经浓缩管(Amicon Ultra-15 centrifugal Filter-10k)浓缩后,与链亲合素-磁珠(Z5482,Promega)在37度孵育1小时,加入200mM NaOH溶液进行碱变性7分钟,打破dsDNA之间的氢键,使得Biotin-ssDNA与链亲合素结合后固定在磁珠上,游离的FITC-ssDNA亚文库则被分离出来。取上清,加入适量100mM盐酸溶液进行中和。
将上述FITC-ssDNA亚文库进行高分辨率的琼脂糖电泳(D615,Takara),电压设定为180V,电泳时间为40分钟,再次分离FITC-ssDNA及其中的污染序列(dsDNA)。
溴化乙锭染色10分钟,紫外灯下切取FITC-ssDNA的目标条带,通过小分子量DNA纯化回收试剂盒(ZP205,Beijing Zoman Biotechnology,Co.,LTD)进行回收。
2.2适配子的富集
为了减少ssDNA文库的损失,第一轮筛选中仅对靶细胞进行孵育。从第二轮开始,正筛以后引入非靶细胞负筛过程。正筛孵育过程与上述一致, 负筛过程中,37℃环境下孵育1小时以后,离心收集上清,弃去与非靶细胞结合的序列。
重复上述过程5-20个循环,为了获得较高亲和力和特异性的适配子,随着筛选轮数的增加,逐渐增加洗涤力度,洗涤次数也从3次增加到5次,洗涤时间从3分钟增加到5分钟。
筛选过程中每隔几个循环使用流式细胞仪对富集情况进行分析。将靶细胞和非靶细胞消化下来,用富集文库与其进行37度孵育1小时。PBS洗三次后,加入400ul PBS重悬细胞,流式细胞仪分析荧光强度。待荧光强度达到一定程度时,终止筛选。
2.3适配子的测序
将最后富集到的文库PCR扩增成双链DNA,使用克隆试剂盒(Invitrogen,cat.no.K4500-01)进行克隆,经蓝白斑筛选,随机挑选多个克隆进行测序。
根据测序结果使用RNA structure软件和DNAMAN软件进行一级序列和二级结构的分析。根据分析结果进行截断和鉴定。最后挑选出三条候选适配子,一级序列为:
2.4 HPLC分析
将细胞裂解,取0.2mL细胞裂解液,加入50ul I.S.(2ug/ml),和1.2mL乙酸乙酯溶液,震荡后离心5分钟。取有机溶液1mL水浴干燥,0.1mL甲醇复溶,4度离心10分钟。取上清液(10ul)进样分析。
色谱条件:Zorbax Extend-C18色谱柱(Agilent Technologies,U.S.A.),ODS预柱(Security Guard,U.S.A.),柱温30度,流动相为甲 醇/乙腈/水(15/20/65,v/v/v%),流速1.0ml/min,检测波长218nm。
含有适配子的雷公藤甲素衍生物的制备
实施例1
适配子-琥珀酰亚胺烷基酰基-多肽连接键-对氨基苯甲氧羰基-雷公藤甲素结构的合成(简称“目的产物6”-多肽以缬氨酸和瓜氨酸连接的二肽)。
1.中间产物3的合成
在氮气氛下,将82.9mg雷公藤甲素2(0.23mmol)溶于干燥的二氯甲烷中,加入几滴吡啶,降温至-50℃,滴加273.8mg(1.27mmol)的对硝基苯甲氧酰氯1的二氯甲烷溶液,滴加完毕后在-50℃搅拌,12小时后,反应混合物用二氯甲烷稀释,所得物质用0.5N的硫酸氢钾溶液洗涤,饱和食盐水洗涤,收集有机相用无水硫酸钠干燥,过滤之后,减压回收溶剂,剩余物用柱色谱分离得到产物3。MS理论计算525.16,实际检测到525.21。
2.中间产物5的合成
氮气氛下将47.3mg化合物3(0.09mmol)溶解于干燥的四氢呋喃-二氯甲烷1∶1的混合溶剂5mL中,加入51.5mg化合物4(0.09mmol)和25mg N,N-二异丙基乙基胺(0.19mmol),室温下搅拌48小时,加入乙酸乙酯,有机相用10%柠檬酸溶液洗涤,饱和食盐水洗涤,水洗,无水硫酸钠干燥,过滤,减压回收溶剂,剩余物用柱色谱分离得到产物5。MS理论计算958.43,实际检测到958.38。
3.目的产物6的合成
将300nmol含硫硫键的LC4溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。将200mmol化合物5溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到目的产物6。MS理论计算16393.38,实际检测到16393.96。
实施例2
适配子-琥珀酰亚胺烷基酰基-多肽连接键-对氨基苯甲醚-雷公藤甲素结构的合成(简称“目的产物10”-多肽以缬氨酸和瓜氨酸连接的二肽)。
1.中间产物7的合成
在氮气氛下将化合物4108.8mg(0.19mmol)溶解于溶解于干燥的DMF中,加入碳酸铯13mg(0.04mmol),三氯乙腈0.2mL(1.9mmol),室温下搅拌,TLC跟踪反应至反应结束,过滤,母液浓缩后柱色谱分离得到中间产物7。MS理论计算715.21,实际检测到715.24。
2.中间产物9的合成
将71.7mg化合物7(0.10mmol)和36.0mg雷公藤甲素2(0.10mmol)悬浮于二氯甲烷中搅拌,冷却到0℃,滴加4.4μL三氟甲磺酸(0.05mmol),TLC跟踪至反应结束,减压蒸去溶剂,柱色谱分离得到产物9。MS理论计算914.44,实际检测到914.38。
将300nmol含硫硫键的LC4溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。将200mmol化合物9溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物10。MS理论计算16349.40,实际检测到16349.97。
实施例3
适配子-丁二酰-多肽连接键-对氨基苯甲氧羰基-雷公藤甲素结构的合成(简称“目的产物15-以缬氨酸和瓜氨酸连接的二肽)。
1.目的产物12的制备
(Fmoc是指芴甲氧羰基保护基)
氮气氛下,将47.3mg化合物3(0.09mmol)溶解于干燥的四氢呋喃-二氯甲烷1∶1的混合溶剂5mL中,加入54.2mg化合物11(0.09mmol)和25mg N,N-二异丙基乙基胺(0.19mmol),室温下搅拌48小时,加入乙酸乙酯,有机相用10%柠檬酸溶液洗涤,饱和食盐水洗涤,水洗,无水硫酸钠干燥,过滤,减压回收溶剂,剩余物用柱色谱分离得到产物12。MS理论计算987.43,实际检测到987.47。
2.中间产物13的制备
在氮气氛下,将29.6mg化合物12(0.03mmol)加入到20%哌啶的DMF 溶液3mL中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,剩余物用柱色谱分离得到产物13。MS理论计算765.36,实际检测到965.42。
3.中间产物14
氮气氛下,将15.3mg(0.02mmol)化合物13溶解于0.2mL二氯甲烷中,往体系中加入琥珀酸酐6.0mg(0.06mmol),DMAP 7.3mg(0.06mmol)。混合物在室温下搅拌过夜,往体系中加入水稀释,用乙酸乙酯萃取(3*10mL),有机相用饱和食盐水洗涤,无水硫酸钠干燥。减压蒸去溶剂,剩余物用柱色谱提纯得到产物14。MS理论计算865.37,实际检测到865.33。
4.目的产物15的制备
将20nmol核酸适配子LC4溶于200μL 0.5M Na2CO3/NaHCO3buffer(pH 9.0),12nmol化合物14溶于200μL DMSO,40nmol DMT-MM溶于200μL DMSO。将三者在0℃混合搅拌12小时后,浓缩用反相高效液相色谱提纯冻干得到产物15。MS理论计算16265.34,实际检测到16265.96。
实施例4
适配子-琥珀酰亚胺烷基酰基-糖苷连接键-取代对羟基苯甲氧羰基N,N’-二甲基乙二胺羰基-雷公藤甲素结构的合成(简称“目的产物21”)。
1.中间产物17的制备
将91.4mg化合物16(0.10mmol)溶于二氯甲烷中,-50℃加入13.2mg N,N’-二甲基乙二胺(0.15mmoB,搅拌,并逐渐升温至室温,TLC跟踪反应至结束,减压蒸去溶剂,剩余物用柱色谱提纯得到产物17。MS理论计算862.33,实际检测到862.37。
2.中间产物18的制备
将78.8mg化合物3(0.15mmol)溶解于二氯甲烷中,-50℃加入86.3mg化合物17(0.10mmol)的二氯甲烷溶液,-50℃搅拌,并逐渐升温至室温,TLC跟踪反应至结束,减压蒸去溶剂,剩余物用柱色谱提纯得到产物18。MS理论计算1248.46,实际检测到1248.55。
3.中间产物19的制备
将37.5mg的化合物20(0.03mmol)溶解于甲醇中,加入含6.5mg氢氧化锂(0.27mmol)的水溶液,室温搅拌,TLC跟踪至反应结束。减压蒸去溶 剂,剩余物用柱色谱分离得到产物19。MS理论计算886.35,实际检测到886.42。
4.中间产物20的制备
将319.3mg化合物19(0.36mmol)和123.3mg 6-(马来酰亚胺基)己酸琥珀酰亚胺酯(MC-OSu)(0.40mmol)溶于5mLN-甲基吡咯烷酮(NMP)中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,往得到的油状物加入乙醚,有沉淀析出,过滤,得到的滤饼用乙醚洗涤得到产物20。MS理论计算1079.42,实际检测到1079.65。
5.目的产物21的制备
将300nmol含硫硫键的LC5溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。200mmol化合物20溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物21。MS理论 计算17784.38,实际检测到17784.87。
实施例5
适配子-琥珀酰亚胺烷基酰基-硫硫键-对巯基苯甲氧羰基-雷公藤甲素结构的合成(简称“目的产物-27”)。
1.中间产物23的制备
将38.9mg化合物22(0.10mmol)溶于二氯甲烷中,滴加16.8mg对巯基苯甲醇(0.12mmol)的二氯甲烷溶液,室温搅拌,TLC跟踪至反应结束,减压蒸去溶剂,剩余物用柱色谱分离得到产物23。MS理论计算437.11,实际检测到437.23。
2.中间产物24的制备
将52.6mg化合物3(0.1mmol)溶解于二氯甲烷中,-50℃加入65.6mg化合物23(0.15mmol)的二氯甲烷溶液,-50℃搅拌,并逐渐升温至室温,TLC跟踪反应至结束,减压蒸去溶剂,剩余物用柱色谱提纯得到产物24。MS理论计算823.25,实际检测到823.33。
3.中间产物25的制备
将24.7mg化合物24(0.03mmol)加入到20%哌啶的DMF溶液3mL中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,剩余物用柱色谱分离得到产物25。MS理论计算601.18,实际检测到601.20。
4.中间产物26的制备
将216.6mg化合物25(0.36mmol)和123.3mg MC-OSu(0.40mmol)溶于5mL NMP中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,往得到的油状物加入乙醚,有沉淀析出,过滤,得到的滤饼用乙醚洗涤得到产物26。MS理论计算794.25,实际检测到794.30。
5.目的产物27的制备
将300nmol含硫硫键的LC5溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。化200mmol合物26溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物27。MS理论计算17499.21,实际检测到17499.94。
实施例6
适配子-琥珀酰亚胺烷基酰基-硫硫键-取代巯基丁酰-雷公藤甲素结构的合成(简称“目的产物32)。
1.中间产物29的制备
将10.4g(28.8mmol)雷公藤甲素2,9.5g二环己基碳二亚胺(46mmol),7.4g化合物28(29mmol)和1.0g DMAP(8.2mmol)溶于二氯甲烷中,室温搅拌,TLC跟踪至反应结束,冷却到0℃过滤除去DCU,滤液减压蒸去溶剂,剩余物用柱色谱提纯得到产物29。MS理论计算599.20,实际检测到599.24。
2.中间产物30的制备
将4.4g化合物29(7.4mmol)和0.6g 2-巯基乙胺(7.8mmol)溶于DMF中,室温搅拌,TLC跟踪至原料点基本消失,减压蒸去溶剂,剩余物用柱色谱提纯得到产物30。MS理论计算565.22,实际检测到565.24。
3.中间产物31的制备
将203.7mg化合物25(0.36mmol)和123.3mg MC-OSu(0.40mmol)溶于5mL NMP中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,往得到的油状物加入乙醚,有沉淀析出,过滤,得到的滤饼用乙醚洗涤得到产物31。MS理论计算758.29,实际检测到758.37。
4.目的产物32的制备
将300nmol含硫硫键的LC5溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。200mmol化合物31溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物32。MS理论计算17464.25,实际检测到17463.97。
实施例7
适配子-琥珀酰亚胺烷基酰基-硫硫键-巯基乙氧羰基-雷公藤甲素结构的合成(简称“目的产物36”)。
1.中间产物33
将30.8mg(0.0854mmol)雷公藤甲素2溶于二氯甲烷中,往体系中加入50.7mg三光气(0.171mmol),滴加55mL吡啶(0.68mmol),室温搅拌30分钟后减压蒸去溶剂。加入二氯甲烷溶解,加入48.0mg吡啶二硫基乙醇(0.256mmol),室温搅拌过夜,加入饱和氯化铵溶液淬灭反应,二氯甲烷萃取,有机相用无水硫酸镁干燥。过滤,减压蒸去溶剂。剩余物用柱色谱提纯得到产物33。MS理论计算573.15,实际检测到573.18。
2.中间产物34
将4.2g化合物33(7.4mmol)和0.6g 2-巯基乙胺(7.8mmol)溶于DMF中,室温搅拌,TLC跟踪至原料点基本消失,减压蒸去溶剂,剩余物用柱色谱提纯得到产物34。MS理论计算539.16,实际检测到539.21。
3.中间产物35的制备
将194.3mg化合物34(0.36mmol)和123.3mg MC-OSu(0.40mmol)溶于5mL NMP中,室温搅拌,TLC跟踪至反应结束。减压蒸去溶剂,往得到的油状物加入乙醚,有沉淀析出,过滤,得到的滤饼用乙醚洗涤得到产物35。MS理论计算732.24,实际检测到732.31。
4.目的产物36的制备
将300nmol含硫硫键的LC6溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。将200mmol化合物35溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物36。MS理论计算17626.14,实际检测到17626.87。
实施例8
适配子-琥珀酰亚胺烷基胺二酰基-多肽连接键-对氨基苯甲氧羰基-雷公藤甲素结构的合成(简称“目的产物39”,以缬氨酸和瓜氨酸连接的二肽)。
1.中间产物38的制备
将44.1mg化合物13(0.0576mmol),19.0mg二环己基碳二亚胺(0.092mmol),7.4mg化合物37(0.0288mmol)和2.0g DMAP(0.0164mmol)溶于二氯甲烷中,室温搅拌,TLC跟踪至反应结束,冷却到0℃过滤除去DCU,滤液减压蒸去溶剂,剩余物用柱色谱提纯得到产物38。MS理论计算1750.77,实际检测到1750.86。
2.目的产物39的制备
将300nmol含硫硫键的LC6溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。将400mmol化合物38溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到产物39。MS理论计算18644.73,实际检测到18644.35。
实施例9
适配子-丁二酰-雷公藤甲素结构的合成(简称“目的产物41”)。
目标产物41的合成
将1.6eq的LC6溶于pH=9.0的碳酸钠和碳酸氢钠的缓冲液中,一次加入溶于DMSO中的1.0eq.的40和溶于DMSO中的1.0eq.的DMT-MM,反应体系保持在室温条件下反应12小时,反应结束后,用RP HPLC纯化粗品后得到目标产物41。MS理论计算17336.06,实际检测到17336.87。
实施例10
适配子-羰基乙烯基-雷公藤甲素结构的合成(简称“目的产物43”)。
目标产物43的合成
将1.6eq.的LC6溶于pH=9.0的碳酸钠和碳酸氢钠的缓冲液中,一次加入溶于DMSO中1.0eq.的42和溶于DMSO中1.0eq.的DMT-MM,反应体系保持在室温条件下反应12小时,反应结束后,用RP HPLC纯化粗品后得到目标产物43。MS理论计算17304.11,实际检测到17304.93。
实施例11
适配子-琥珀酰亚胺烷基酰基-多肽连接键-对氨基苯甲氧羰基-雷公藤甲素18-位偶合物的合成(简称“目的产物48”)
1.中间产物45的合成
氮气氛下将109.17mg 44(0.23mmol)溶于干燥的四氢呋喃中,在-78℃缓慢加入稍微过量的LDA,在此温度下搅拌一个小时后,滴加273.8mg化合物1(1.27mmol)的二氯甲烷溶液,滴加完毕后在-50℃搅拌,12小时后,反应混合物用二氯甲烷稀释,所得物质用0.5N的硫酸氢钾溶液洗涤,饱和食盐水洗涤,收集有机相用无水硫酸钠干燥,过滤之后,减压回收溶剂,剩余物用柱色谱分离得到产物45。MS理论计算653.27,实际检测到653.31。
2.中间产物46的合成
氮气氛下将58.8mg化合物45(0.09mmol)溶解于干燥的四氢呋喃-二氯甲烷1∶1的混合溶剂5mL中,加入51.5mg化合物4(0.09mmol)和25mg N,N-二异丙基乙基胺(0.19mmol),室温下搅拌48小时,加入乙酸乙酯,有机相用10%柠檬酸溶液洗涤,饱和食盐水洗涤,水洗,无水硫酸钠干燥,过滤,减压回收溶剂,剩余物用柱色谱分离得到产物46.MS理论计算1072.52,实际检测到1072.65。
2.中间产物47的合成
将107.3mg的化合物46(0.10mmol)溶于干燥的四氢呋喃中,在室温条件下,加入31.3mg的四丁基氟化铵(0.12mmol),常温搅拌反应3小时后,浓缩,再用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥,过滤,蒸干,剩余物用柱色谱分离得到产物47.MS理论计算958.43,实际检测到958.56。
4.目标产物48的合成
将300nmol含硫硫键的LC4溶解于PBS缓冲溶液(pH 7.4),加入三羧乙基膦还原2小时,用G-25 Sephadex size-exclusion column除去过量的三羧乙基膦。将200mmol化合物47溶于50mL DMF中,和洗脱物一起在冰浴下培育12小时后,浓缩用反相高效液相色谱提纯冻干得到目的产物48。MS理论计算16393.38,实际检测到16393.96。
实验例1雷公藤甲素衍生物-骨肉瘤适配子偶合物体内活性考察
1.实验材料:
实验药品及试剂:雷公藤甲素(以下简称“TP”),雷公藤甲素衍生物(实施例1中制备的中间产物5,以下简称“TP-D”,雷公藤甲素-适配子偶合物(采用实施例1目标产物化合物6,以下简称“A-TP-D”),DMEM培 养基,胎牛血清,青霉素-链霉素,基质胶,无水乙醇,生理盐水,乙酸乙酯,乙腈等。
细胞:人成骨细胞MG63。
实验动物:雌性裸鼠。
实验仪器:超净工作台,CO2培养箱,倒置显微镜,高速离心机,超速离心机,真空干燥器,高效液相色谱仪。
2.试验方法:
2.1骨肉瘤模型构建:
2.1.1人成骨细胞MG63培养:人成骨细胞MG63购自中国科学院上海细胞库。细胞用含有10%的胎牛血清(购自sigma公司)和1.2ml/1000ml的青霉素-链霉素(购自sigma公司)的DMEM培养基(购自sigma公司),于5%CO2,37℃的孵箱中进行培养。倒置显微镜对细胞进行形态观察和计数,所有的实验取对数生长阶段的细胞进行。
2.1.2人成骨细胞MG63原位荷瘤裸鼠模型的建立:雌性裸小鼠由香港浸会大学中医药学院动物房提供并饲养。取对数生长期的MG63细胞,用50∶50的基质胶和无血清培养基重悬至浓度为2×107个/ml。选取体重20g左右,6-8周龄的雌性裸小鼠,于右侧背部皮下注射100μl细胞悬液进行接种。每天用双向游标卡尺测量肿瘤的大小,个体的肿瘤体积公式如下:V=(长x宽2)/2,长取肿瘤最长的直径,宽取垂直于长的最短的直径,肿瘤生长至200mm3时,进行后续试验。
2.2实验动物分组及给药
2.2.1受试药物配置:雷公藤甲素、雷公藤甲素衍生物(中间产物5)、雷公藤甲素-骨肉瘤适配子偶合物(目标产物6)。分别称取雷公藤甲素和中间产物5溶于50∶50乙醇/生理盐水溶液中,配置成浓度为0.6mg/ml的药物溶液,置于4℃冰箱备用;
称取目标产物6溶于50∶50乙醇/生理盐水中,配置成浓度为22.0mg/ml的药物溶液,置于4℃冰箱备用。
2.2.2实验分组及处理:肿瘤生长至200mm3时,将30只荷瘤小鼠,随机分为A,B组。其中A组按0.6mg/kg剂量尾静脉注射雷公藤甲素的乙醇/ 生理盐水溶液;B组按22.0mg/kg剂量尾静脉注射目标产物6的乙醇/生理盐水溶液。给药体积均为1ml/kg。分别于0.5,2,6,12,和24h时,对每组3只老鼠采用腹腔注射戊巴比妥钠麻醉,采用心脏穿刺取血,摘取心、肝、脾、肺、肾、实体瘤等组织。血样于4℃以3000rpm离心20min,取血清备用;心、肝、脾、肺、肾、实体瘤等组织于-80℃保存备用。
2.3 HPLC检测组织中雷公藤甲素/雷公藤甲素-骨肉瘤适配子偶合物含量
称取一定重量的组织(心,肝,脾,肺,肾,肿瘤),加入一定比例的PH7.5PBS溶液,匀浆。取0.2ml匀浆到eppendorf管中,1.2ml乙酸乙酯涡流萃取3分钟,8000rpm离心5min。取上层有机溶液层(1ml)转移到另一个eppendorf管中真空干燥处理,残留物再溶入0.1ml乙醇,4℃下20000rpm离心10min。取上清液(10μl)注入安捷伦1100型高效液相色谱仪。色谱条件为:分离柱Symmetry ShieldTM RP18 column(4.6mm×250mm,5um,),保护柱ODS guid column(3.9mm×20mm,5um);流动相:乙腈-水(23∶77);流速1.0mL/min;柱温35℃;检测波长219nm;进样体积20.0uL。
2.4雷公藤甲素-骨肉瘤适配子偶合物体内稳定性考察
取2.2.2中B组样品。通过2.3中已建立起方法的HPLC分别检测每个时间点各组织中游离的雷公藤甲素衍生物和雷公藤甲素衍生物-骨肉瘤适配子的浓度,并计算出释放率(释放率=(雷公藤甲素浓度)/(雷公藤甲素浓度+雷公藤甲素-骨肉瘤适配子浓度)),监测-定量考察药物在组织内的稳定性。
2.5雷公藤甲素-骨肉瘤适配子偶合物组织分布考察
取2.2.2中A,B组样品。通过2.3中已建立好的HPLC方法进行监测-定量考察药物在正常器官、肿瘤组织的分布情况。
2.6雷公藤甲素-骨肉瘤适配子偶合物体内抗肿瘤活性研究:肿瘤生长至200mm3时,将24只荷瘤小鼠平均分成4组(每组n=6):C:PBS组;D:游离雷公藤甲素衍生物组(TP-D,中间产物5);E:游离雷公藤甲素组(TP);F:雷公藤甲素-骨肉瘤适配子偶合物组(A-TP-D,目标产物6)。PBS组作为该实验的空白对照组。其他三组分别尾静脉注射相应的药物,D、E组药物剂量为0.6mg/kg,F组药物剂量为22.0mg/kg一周给药两次。继续每四 天测量一次肿瘤大小和体积。给药后第一天,每只裸鼠腹腔注射3%戊巴比妥钠进行麻醉,心脏穿刺收集血液样品,检测血清中的化疗抵抗相关因子NF-κB的表达水平。给药8周后,处死所有裸鼠,分离肿瘤组织,-80℃保存备用。
2.7统计学分析
实验数据采用平均值±标准差(SD)表示,数据处理采用Graphpad Prism6.0统计软件。差异显著性检验采用t检验和方差分析法进行。
3.实验结果:
3.1.雷公藤甲素-骨肉瘤适配子偶合物在体内各组织中的释放率
各时间点雷公藤甲素-骨肉瘤适配子偶合物在各组织中的解释放率由HPLC法确定。考察结果如下。雷公藤甲素-骨肉瘤适配子偶合物经过24小时后在血浆及心,肝,脾,肺,肾等正常组织中几乎不解离或解离得很少;而在肿瘤组织中,雷公藤甲素-骨肉瘤适配子偶合物大量解离,并且随着时间的累积,解离度不断增加。以上结果证明了用于连接雷公藤甲素和骨肉瘤适配子的酸性敏感的烯醚键在体内也有作用。偶合物在正常组织中不断裂,保护了C-14位基团,减小毒性;在肿瘤组织中断裂,释放出雷公藤甲素衍生物,发挥治疗作用参见表1及图1。
表1
3.2.雷公藤甲素-骨肉瘤适配子偶合物体内选择性分布
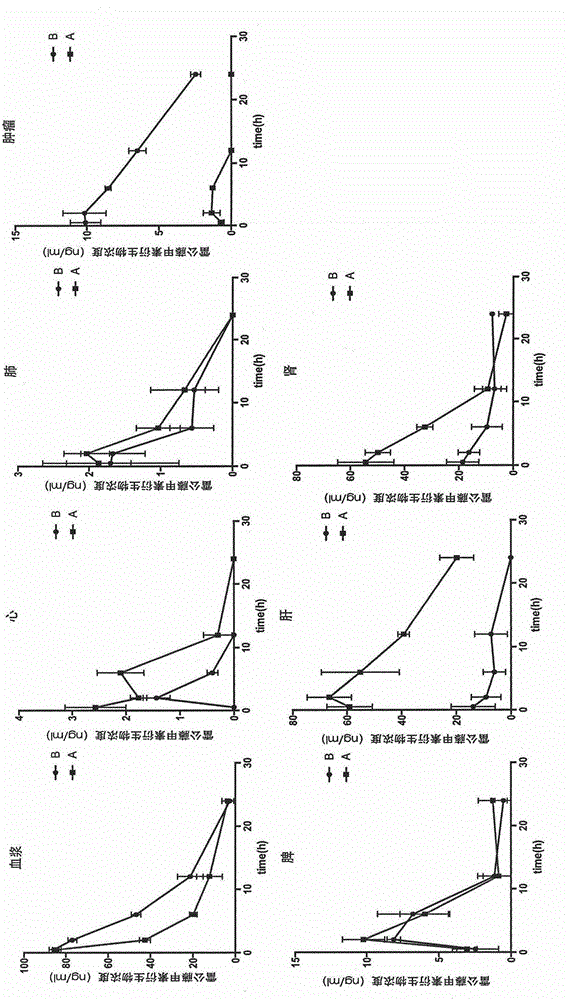
采用HPLC定量检测药物在正常器官、肿瘤组织的分布情况。考察结果见图2。给药后0.5小时内,对照组A和实验组B在给药0.5小时内血药浓度迅速升高到顶峰,但是实验组B在血中消除速度要比对照组A慢。实验组B和对照组A相比,药物在心脏,脾脏,肺部三个组织部位的分布趋势在考察全时间段内都相似(都换算为雷公藤甲素的浓度)。在给药后0.5-2小时内,雷公藤甲素衍生物迅速向心,脾,肺三个组织分布,给药后4小时开始消除。相反地,实验组B和对照组A相比,药物在肝脏,肾脏和肿瘤三个组织部位的分布趋势有很大的不同(都换算为雷公藤甲素的浓度)。实验组B分布在肝,肾组织的雷公藤甲素的浓度明显要比对照组A低,而分布在肿瘤部位的雷公藤甲素的浓度要比对照组A高出很多。以上结果说明骨肉瘤适配子的修饰作用能促使雷公藤甲素选择性向肿瘤部位积聚,而避免了在其它组织,尤其是肝肾组织累积。参见表2及图2。
表2
3.3.雷公藤甲素-骨肉瘤适配子偶合物体内抗肿瘤活性研究:
3.3.1肿瘤体积的测量:如图3所示,PBS组裸鼠肿瘤体积呈进行性增大,中间产物5(TP-D)组在开始注射药物后肿瘤体积呈缓慢减小趋势,与游离雷公藤甲素组(TP)组相比,肿瘤体积减小更明显,目标产物6(A-TP-D)组肿瘤体积减小最快。
3.3.1裸鼠血清中NF-κB的mRNA表达水平:如图4所示,PBS组裸鼠血清中NF-κB的mRNA表达水平随着时间的延长逐渐升高,游离雷公藤甲素衍生物组(TP-D)组和游离雷公藤甲素组(TP)组在第三次注射药物后NF-κB的mRNA表达开始下降,且两组在同一时间点无明显差别。雷公藤甲素衍生物-骨肉瘤适配子偶合物组(A-TP-D)组血清中NF-κB的mRNA水平在第三次注射药物后达到最大,但较TP-D和TP组水平低,此后NF-κB的mRNA水平开始下降,且在同一时间点较TP-D和TP组均明显降低。
4.实验结论:
雷公藤甲素衍生物-骨肉瘤适配子偶合物具有体内骨肉瘤选择性靶向作用;雷公藤甲素衍生物-骨肉瘤适配子偶合物在偶合物在正常组织中不断裂,毒性小;雷公藤甲素衍生物-适配子偶合物在体内抗肿瘤活性高于雷公藤甲素衍生物和雷公藤甲素。
- 还没有人留言评论。精彩留言会获得点赞!