7‑乙基‑10‑羟基喜树碱‑聚合物偶联药物及其纳米制剂制备方法与流程
本发明涉及7-乙基-10-羟基喜树碱偶联聚乳酸或者聚乳酸-羟基乙酸共聚物的前药,前药的制备、其纳米制剂的构建及体内外抗肿瘤作用。
背景技术:
7-乙基-10-羟基喜树碱(SN38)是喜树碱的衍生物,具有极强的抑制肿瘤细胞增殖的活性。主要通过与拓扑异构酶I及DNA的复合物(TOPcc)结合,从而抑制DNA的解链和肿瘤细胞的增殖。由于SN38分子不溶于水,也不溶于药剂学可以接受的溶剂(如吐温80、聚氧乙烯蓖麻油等),导致不能直接通过静脉注射的方式用于临床。此外SN38一旦进入血液循环系统,很容易被水解并与血清白蛋白结合从而失去活性。美国FDA于1997年批准了水溶性的SN38衍生物伊立替康(Irinotecan,CPT-11)用于临床各种癌症的治疗。但是临床应用该药物具有非常大缺陷:i)伊立替康在体内转化成具有抗癌作用的SN38需要羧酸酯酶的酶解,但其在体内的转化率不到8%,导致生物利用率极低;ii)伊立替康在血液循环中仍然容易水解而导致失去活性;iii)伊立替康的代谢产物SN38在肠道残留,产生较大的副作用如血性腹泻,限制了药物剂量的提高。综上所述,采用SN38的核心结构,对其高效地利用,并降低该药物分子的临床应用副作用具有非常重要意义。
针对上述科学问题,目前已开发了各种纳米尺寸的药物输送系统来充分利用SN38治疗肿瘤。如日本科学家开发了聚二乙醇聚谷氨酸共聚物用于共价偶联SN38分子,具有两亲性的高聚物在水相中自组装形成直径约为20nm的颗粒,在皮下动物肿瘤模型中取得了有效的抑瘤效果(Koizumi et al.,Novel SN-38–Incorporating Polymeric Micelles,NK012,Eradicate Vascular Endothelial Growth Factor–Secreting Bulky Tumors,Cancer Research,2006,66,10048-10056)。相对CPT-11小分子药物,包裹有SN38的纳米药物进入体内循环后,载体可以有效保护药物分子,从而避免其降解或者失活。此外,肿瘤组织特殊的生物学特征,使得纳米材料通过实体瘤的高通透和滞留效应(EPR效应)对其具有被动靶向功能,提高药物的利用率。再者药物包裹于载体后,可有效降低血液中游离的药物的浓度,从而降低药物毒性。
本发明中,采用无毒、无刺激性、生物相容性好,并且可生物降解的聚乳酸(PLA)、聚乳酸-羟基乙酸共聚物(PLGA),对SN38进行分子改性,使其可与两亲性高分子如聚乙二醇-聚乳酸(mPEG-PLA)或者聚乙二醇-聚乳酸-羟基乙酸共聚物(mPEG-PLGA)自组装,形成纳米粒,实现药物在血液中实现长循环的目的,提高抗癌效果。
技术实现要素:
本发明提供了可生物降解的聚乳酸(PLA)或者聚乳酸-羟基乙酸共聚物(PLGA)共价偶联SN38上C-10或者C-20上的羟基,生成SN38-聚合物偶联前药,其偶联前药如式I所示,
其中,R1和R2为氢或具有如下结构,为端羧酸聚乳酸或者端羧基聚乳酸-羟基乙
酸共聚物,且R1≠R2,
m为1-5,表示引发剂的乙二醇数;n为0-18,表示两种聚合物中羧酸烷烃碳数,
虚线表示为此处为单键或双键。x为7-280,表示乳酸聚合度,y为8-100,表示羟基乙酸聚合度。
本发明中,所述端羧酸聚合物中,羧酸烷烃碳链为脂肪二酸或者不饱和脂肪二酸;
作为优选,所述n为1-6,进一步优选n=2;
更为优选地,端羧酸聚合物选自聚乳酸(600)-丁二酸、聚乳酸(1200)-丁二酸、聚乳酸(2600)-丁二酸、聚乳酸(5100)-丁二酸、聚乳酸(10000)-丁二酸、聚乳酸(20000)-丁二酸、羧酸聚乳酸-羟基乙酸(5100)-丁二酸。
更为优选地,端羧酸聚合物选自聚乳酸(600)-丁二酸、聚乳酸(1200)-丁二酸、聚乳酸(2600)-丁二酸、聚乳酸(5100)-丁二酸;
本发明中,所述端羧酸聚乳酸分量在500-20000,端羧酸聚乳酸-羟基乙酸共聚物分子量在1000-25000。
本发明提供了式I所示的偶联前药的制备方法,包括:其中式I制备方法:
在缩合剂存在的情况下,端羧酸聚合物和SN38进行酯化反应,分离纯化得到式I所示的聚合物-SN38偶联前药。
优选的,当R2=H时,所述式I的制备方法为:在SN38上C-10上引入保护基,在缩合剂存在的情况下,所述聚合物与SN38上C-20上羟基进行酯化反应,脱去上一步引入的保护基,分离纯化得到SN38-聚合物偶联前药。
本发明所提供的所有制备过程,所述的缩合剂均可选为N,N’-二环乙基碳二亚胺(DCC)、1-(3-二甲氨基)-3-乙基碳二亚胺(EDC)或N,N’-二异丙基碳酰亚胺(DIPC)。
所述的反应中温度为10-80℃,优选为48℃。
所述的反应中反应溶剂可以为二氯甲烷,DMF(二甲基甲酰胺)、DMSO(二甲基亚砜)或者上述溶剂两种或者三种的混合液。
本发明也提供了SN38-聚合物纳米制剂制备,包括:
经酯化反应得到的SN38-聚合物偶联前药和两亲性高分子材料溶解于有机溶剂中,在一定比例下滴加到水溶液中,利用旋转蒸发仪除去有机溶剂后得到聚乳酸-SN38纳米制剂。
所述两亲性高分子材料为聚乙二醇和脂肪族聚酯的共聚物,其中脂肪族聚酯优选为聚乳酸和聚乳酸-羟基乙酸共聚物,即分别表示为如聚乙二醇-聚乳酸(mPEG-PLA)、聚乙二醇-聚乳酸-羟基乙酸共聚物(mPEG-PLGA)。脂肪族聚酯更优选为聚乳酸。
所述聚乙二醇的数均分子量为2000~8000,脂肪族聚酯指乳酸-羟基乙酸(PLGA)共聚物或聚乳酸共聚物(PLA),分子量2000~20000。
所述SN38-聚合物偶联前药以0.5~2mg/ml,两亲性高分子材料以5~50mg/ml溶解在有机溶剂如四氢呋喃、乙腈、丙酮、乙醇、甲醇中的一种或者两两混合有机溶剂中。混合均匀后,滴加入水中的SN38-聚合物偶联前药浓度为0.06~0.5mg/ml,两亲性高分子材料的浓度为0.625~6.25mg/ml.
本发明提供了所述SN38-聚合物纳米制剂的粒径和扫描电镜。所述前药纳米制剂的粒径为20~50nm。
本发明还提供了SN38-聚合物纳米制剂在磷酸缓冲液、37℃条件下的药物释放。释放实验表明,在该条件下,纳米制剂中抗肿瘤活性分子SN38被缓慢释放,且不同分子量的聚乳酸对释放速度有很大程度上的影响。
内含SN38的纳米载体在血液中实现长循环的目的,到达肿瘤组织后缓慢释放出抗癌活性分子SN38,显著提高抗癌效果。
另外所述偶联前药纳米制剂中,构建SN38前药及制备纳米制剂所使用的高分子材料都是美国FDA批准用于临床的,因此有望提高药物的临床转化的前景。此外,疏水的SN38在mPEG-PLA或者mPEG-PLGA沉淀形成纳米粒的过程中,使得药物分子处于纳米粒的疏水内核,在血液循环中有效保护药物,从而降低毒性,提高药物利用率。
本发明还提供不同分子量聚乳酸和SN38制备的SN38-聚乳酸纳米制剂细胞毒性实验和动物药效学实验。细胞毒性实验表明所述纳米制剂对肿瘤细胞的毒性不亚于SN38单分子直接用药。动物皮下肝癌模型进行对所述纳米制剂的药效学评价,实验表明,对照生理盐水组,实验组肿瘤体积缩小了8-120倍,对照伊立替康组,肿瘤缩小2-29倍。SN38-聚合物偶联纳米药物治疗组生存时间可以延长3倍。
与现有技术相比,本发明的有益效果体现在:
(1)与临床用药伊立替康极低的酶促水解产率相比,本发明中的SN38-聚合物偶联药物包裹于疏水内核,在生理环境下,既有效降低水解导致的失活,又能通过载体的降解从而缓慢均衡释放具有抗癌活性的SN38分子。
(2)本发明通过不同分子量的聚乳酸调控SN38的释放速度,在减缓药物释放的同时,提高药物在体循环中的稳定性。
(3)本发明中构建SN38-聚合物偶联前药及制备纳米制剂时所使用的高分子材料都是美国FDA批准用于临床的,在体内都可被吸收以及自行进行生物降解的,安全性高,符合临床药物制备要求,符合大规模生产的要求具备良好的市场前景。
(4)从图9可以得出制备得到的纳米药物的颗粒直径主要分布在20-50nm,满足纳米颗粒通过实体瘤的高通透和滞留效应(EPR效应),增加其被动靶向功能。
本发明中的缩略语,mPLA(600)-SA、mPLA(1200)-SA、mPLA(2600)-SA、mPLA(5100)-SA、mPLA(10000)-SA和mPLA(20000)-SA分别表示分子量不同的端丁二酸聚乳酸;mPLA(5000)-SA表示端丁二酸聚乳酸-羟基乙酸共聚物;EDC表示1-(3-二甲氨基丙基)-3-乙基碳二亚胺的盐酸盐;(Boc)2O表示二羰基二叔丁酯;TFA表示三氟乙酸;mPEG(5k)-PLA(8K)表示聚乙二醇-聚乳酸共聚物;DCM表示二氯甲烷;MeOH表示甲醇。
附图说明
图1为实施例1SN38-PLA(600)偶联前药1的合成路线;
图2为实施例2SN38-PLA(1200)偶联前药2的合成路线;
图3为实施例3SN38-PLA(2600)偶联前药3的合成路线;
图4为实施例4SN38-PLA(5100)偶联前药4的合成路线;
图5为实施例5SN38-PLA(10000)偶联前药5的合成路线;
图6为实施例6SN38-PLA(20000)偶联前药6的合成路线;
图7为实施例7SN38-PLA(5100)(C-20)偶联前药7的合成路线;
图8为实施例8SN38-PLGA(5000)前药8的合成路线;
图9为实施例9-12SN38-PLA纳米制剂的粒径分布;
图10-13实施例9-12SN38-PLA纳米制剂的透射电镜;
图14为实施例13SN38-PLA纳米制剂的体外释放;
图15为实施例15SN38-PLA纳米制剂的体内抗肿瘤药效评价。
具体实施方式
下面的实施例用来进一步说明本发明,但是这不意味着对本发明的任何限制。
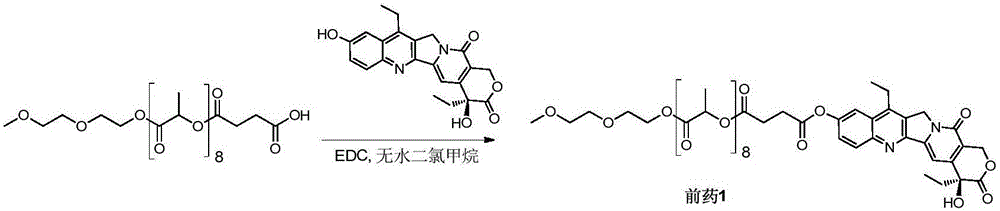
实施例1 SN38-聚乳酸偶联前药1的合成(图1)
在100mL圆底烧瓶中加入mPLA(600)-SA(207mg,0.26mmol)和SN38(100mg,0.26mmol),溶解在10mL无水二氯甲烷中,再加入EDC(59mg,0.38mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物1(102mg,34%)。
前药1的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.00-1.01(t,3H),1.38-1.42(t,3H),1.58(s,24H),1.88-1.92(q,2H),2.89-2.92(t,2H),2.98-3.03(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.53-3.56(t,2H),3.61-3.64(t,2H),3.68-3.70(t,2H),4.25-4.33(m,2H),5.15-5.22(m,8H),5.27(s,2H),5.36(s,1H),5.74-5.78(d,1H,J=16.4),7.57-7.59(t,1H),7.65(s,1H),7.85-7.85(d,1H,J=2.4),8.22-8.25(d,1H,J=9.2).
实施例2 SN38-聚乳酸偶联前药2的合成(图2)
在100mL圆底烧瓶中加入mPLA(1200)-SA(357mg,0.26mmol)和SN38(100mg,0.26mmol),溶解在13mL无水二氯甲烷中,再加入EDC(59mg,0.38mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物2(185mg,41%)。
前药2的1H NMR核磁数据以及质谱数据如下:
1HNMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.38-1.42(t,3H),1.57-1.61(m,51H),1.88-1.92(q,2H),2.89-2.94(m,2H),2.98-3.07(m,2H),3.13-3.18(q,2H),3.38(s,3H),3.54-3.56(q,2H),3.63-3.65(q,2H),3.68-3.70(t,2H),4.25-4.33(m,2H),5.15-5.22(m,17H),5.27(s,2H),5.33(s,1H),5.73-5.77(d,1H,J=16.0),7.55-7.58(q,1H),7.66(s,1H),7.84-7.84(d,1H,J=2.4),8.22-8.24(d,1H,J=9.2).
实施例3 SN38-聚乳酸偶联前药3的合成(图3)
在100mL圆底烧瓶中加入mPLA(2600)-SA(591mg,0.20mmol)和SN38(79mg,0.20mmol),溶解在15mL无水二氯甲烷中,再加入EDC(47mg,0.30mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物3(320mg,48%)。
前药3的1H NMR核磁数据以及质谱数据如下:
1HNMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.38-1.42(t,3H),1.53-1.61(m,108H),1.86-1.92(m,2H),2.89-2.92(m,2H),2.98-3.03(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.53-3.56(q,2H),3.63-3.65(t,2H),3.68-3.70(t,2H),4.23-4.34(m,2H),5.13-5.19(m,36H),5.27(s,2H),5.33(s,1H),5.74-5.78(d,1H,J=16.0),7.55-7.58(q,1H),7.65(s,1H),7.84-7.85(d,1H,J=2.4),8.22-8.24(d,1H,J=8.8).
实施例4 SN38-聚乳酸偶联前药4的合成(图4)
在100mL圆底烧瓶中加入mPLA(5100)-SA(1.27g,0.26mmol)和SN38(100mg,0.26mmol),溶解在15mL无水二氯甲烷中,再加入EDC(59mg,0.38mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物4(640mg,47%)。
前药4的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.40-1.42(t,3H),1.53-1.61(m,213H),1.88-1.92(m,2H),2.89-2.92(m,2H),2.98-3.07(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.54-3.56(q,2H),3.63-3.65(t,2H),3.68-3.70(t,2H),4.27-4.31(m,2H),5.13-5.13(m,71H),5.27(s,2H),5.33(s,1H),5.73-5.77(d,1H,J=16.4),7.55-7.58(q,1H),7.65(s,1H),7.84-7.85(d,1H,J=2.4),8.22-8.24(d,1H,J=9.2).
实施例5 SN38-聚乳酸偶联前药5的合成(图5)
在100mL圆底烧瓶中加入mPLA(10000)-SA(1.04g,0.13mmol)和SN38(50mg,0.13mmol),溶解在15mL无水二氯甲烷中,再加入EDC(29.5mg,0.19mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物5(497mg,45%)。
前药5的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.40-1.42(t,3H),1.52-1.60(m,324H),1.88-1.92(m,2H),2.89-2.92(m,2H),2.98-3.07(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.54-3.56(q,2H),3.63-3.65(t,2H),3.68-3.70(t,2H),4.27-4.31(m,2H),5.13-5.14(m,108H),5.27(s,2H),5.33(s,1H),5.73-5.76(d,1H,J=12.0),7.55-7.58(q,1H),7.65(s,1H),7.85-7.86(d,1H,J=2.4),8.22-8.24(d,1H,J=8.8).
实施例6 SN38-聚乳酸偶联前药6的合成(图6)
在100mL圆底烧瓶中加入mPLA(20000)-SA(2.59g,0.26mmol)和SN38(50mg,0.13mmol),溶解在18mL无水二氯甲烷中,再加入EDC(29.5mg,0.19mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物6(919mg,43%)。
前药6的1H NMR核磁数据以及质谱数据如下:
1HNMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.38-1.42(t,3H),1.57-1.63(m,822H),1.88-1.92(q,2H),2.89-2.94(m,2H),2.98-3.07(m,2H),3.13-3.18(q,2H),3.38(s,3H),3.53-3.55(q,2H),3.63-3.65(q,2H),3.68-3.70(t,2H),4.25-4.33(m,2H),5.15-5.23(m,274H),5.27(s,2H),5.33(s,1H),5.73-5.77(d,1H,J=17.2),7.55-7.58(q,1H),7.66(s,1H),7.84-7.84(d,1H,J=2.4),8.21-8.23(d,1H,J=9.6).
实施例7 SN38-聚乳酸(C-20)偶联前药7的合成(图7)
在100mL圆底烧瓶中加入(Boc)2O(145mg,0.66mmol)和SN38(200mg,0.51mmol),溶解在15mL无水二氯甲烷中,再加入EDC(118.8mg,0.77mmol)。50℃搅拌4小时,除去反应溶剂后,分别用饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到中间体7a,Boc-SN38(180mg,80%)。
7a的1H NMR核磁数据以及质谱数据如下:
1HNMR(400MHz,CDCl3):δ1.03-1.06(t,3H),1.39-1.42(t,3H),1.62-1.62(S,9H),1.85-1.96(m,2H),3.14-3.19(m,2H),3.79-3.79(s,1H),5.27(s,2H),5.30-5.34(d,1H,J=16),3.74-3.78(d,1H,J=16),7.26-7.27(s,1H),7.66-7.68(d,1H,J=9.2),7.9(s,1H),8.23-8.26(d,1H,J=10.8).
在100mL圆底烧瓶中加入mPLGA(5100)-SA(1.27g,0.26mmol)和Boc-SN38(132.1mg,0.26mmol),溶解在15mL无水二氯甲烷中,再加入EDC(59mg,038mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)得到中间体7b(506mg,37%)后,加入无水二氯甲烷和三氟乙酸(TFA)分别5ml溶解,室温搅拌0.5h,去除溶剂,用柱层析色谱分离纯化(DCM:MeOH=100:1)得到产物7(396mg,80%)。
7b的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.40-1.42(t,3H),1.53-1.62(m,222H),1.88-1.92(m,2H),2.89-2.91(m,2H),2.98-3.07(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.54-3.56(q,2H),3.63-3.65(t,2H),3.68-3.70(t,2H),4.28-4.31(m,2H),5.12-5.13(m,71H),5.28(s,2H),5.33(s,1H),5.73-5.76(d,1H,J=16.0),7.55-7.58(q,1H),7.65(s,1H),7.84-7.85(d,1H,J=2.4),8.22-8.24(d,1H,J=9.2).
前药7的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.02-1.06(t,3H),1.40-1.42(t,3H),1.53-1.61(m,213H),1.88-1.91(m,2H),2.89-2.92(m,2H),2.98-3.07(m,2H),3.13-3.18(q,2H),3.38(s,3H),3.55-3.57(q,2H),3.63-3.65(t,2H),3.68-3.70(t,2H),4.27-4.31(m,2H),5.13-5.13(m,71H),5.27(s,2H),5.33(s,1H),5.73-5.76(d,1H,J=16.4),7.55-7.58(q,1H),7.64(s,1H),7.84-7.85(d,1H,J=2.4),8.22-8.24(d,1H,J=9.0).
实施例8 聚乳酸-羟基乙酸-SN38偶联前药8的合成(图8)
在100mL圆底烧瓶中加入mPLGA(5000)-SA(1.29g,0.26mmol)和SN38(100mg,0.26mmol),溶解在15mL无水二氯甲烷中,再加入EDC(59mg,0.38mmol)。50℃搅拌4小时,除去反应溶剂后,分别用5%柠檬酸,饱和碳酸氢钠,饱和食盐水清洗。有机相用无水硫酸钠干燥,过滤,收集滤液后减压除去溶剂。用柱层析色谱分离纯化(DCM:MeOH=120:1)后得到产物8(624mg,45%)。
前药8的1H NMR核磁数据以及质谱数据如下:
1H NMR(400MHz,CDCl3):δ1.03-1.06(t,3H),1.40-1.42(t,3H),1.53-1.61(m,120H),1.89-1.93(m,2H),2.89-2.92(m,2H),2.99-3.07(m,2H),3.13-3.19(q,2H),3.38(s,3H),3.54-3.56(q,2H),3.63-3.65(t,2H),3.67-3.70(t,2H),4.27-4.31(m,2H),5.13-5.13(m,70H),5.27(s,2H),5.33(s,1H),5.74-5.77(d,1H,J=16.4),7.55-7.58(q,1H),7.65(s,1H),7.84-7.85(d,1H,J=2.4),8.23-8.26(d,1H,J=9.2).
实施例9 前药1纳米药物的制备
将实施例1中纯化得到的1(10mg,活性分子SN38为4.7mg)和mPEG(5k)-PLA(8k)96mg溶解于3mL丙酮中,均匀后滴加到10mL水中,滴加完毕后,减压去除丙酮,即可得到聚乳酸-SN38纳米药物。透射电镜(TEM)观察纳米粒形貌大小如图10所示。
实施例10 前药2纳米药物的制备
将实施例2中纯化得到的2(15mg,活性分子SN38为3.9mg)和mPEG(5k)-PLA(8k)78mg溶解于3mL丙酮中,均匀后滴加到10mL水中,滴加完毕后,减压去除丙酮,即可得到聚乳酸-SN38纳米药物。TEM观察纳米粒形貌大小如图11所示。
实施例11 前药3纳米药物的制备
将实施例3中纯化得到的3(25mg,活性分子SN38为3.5mg)和mPEG(5k)-PLA(8k)70mg溶解于3mL丙酮中,均匀后滴加到10mL水中,滴加完毕后,减压去除丙酮,即可得到聚乳酸-SN38纳米药物。TEM观察纳米粒形貌大小如图12所示。
实施例12 前药4纳米药物的制备
将实施例4中纯化得到的4(48mg,活性分子SN38为3.4mg)和mPEG(5k)-PLA(8k)68mg溶解于3mL丙酮中,均匀后滴加到10mL水中,滴加完毕后,减压去除丙酮,即可得到聚乳酸-SN38纳米药物。TEM观察纳米粒形貌大小如图13所示。
实施例13 9-12纳米药物的体外释放(图14)
将实施例中9-12制备的纳米药物3mL分别置于分子量为14000kDa的透析袋中,置于外界20mL pH为7.4的磷酸缓冲液中,在温度为37℃,转速为150r/min的环境中,分别在2h、4h、8h、12h、24h、48h、72h和120h取出外界磷酸缓冲液,用紫外分光光度计测得SN38含量,从而得到4个纳米药物的相应的体外释放情况。有图11可知,聚乳酸分子量越高,释放地越缓慢。
实施例14 9-12纳米药物体外细胞毒性评价
为评价实施例9-12制备的纳米药物对肝癌肿瘤细胞的杀伤能力,以Hep3B、LM3、7402为例,采用MTT法进行了药效评价,并以伊立替康及SN38作为对照。对各种肿瘤细胞的毒性结果见表1。由表1可见,本发明中的SN38纳米药物与细胞共培养72小时后,其体外抗肿瘤活性均远远优于临床用药伊立替康,并且具有与SN38相类似的效果。
表1.药物培养72小时后细胞成活率的测定(MTT)IC50±SD inμM
实施例15 9-12纳米药物抗肿瘤动物药效评价(图15)
对实施例9-12制备的纳米药物对动物皮下肝癌肿瘤HCC(来源于肝癌病人)进行抑瘤评价。Balb/c裸鼠移植肿瘤2周后,每隔三天一次尾静脉给药,总共三次:生理盐水,伊立替康(17.25mg/kg),纳米药物5-8(15mg/kg),总共6组。以第一次给药为0天,每隔三天测量瘤体体积变化进行结果统计。对皮下肝癌瘤的药效评价结果见图12。由图可知,实例9-12中的纳米药物,相对于临床伊立替康对抑制肿瘤生长具非常显著的效果,且实例11和12的效果优于实例9和10中的纳米药物。在治疗一个月后,纳米药物11和12用药组的肿瘤以基本消除。
- 还没有人留言评论。精彩留言会获得点赞!