作为用于癌症治疗和诊断之靶标的磷酸甘油酸激酶1的蛋白激酶活性的制作方法
发明背景
本申请要求2015年1月5日提交的美国临时申请号62/099,899的优先权权益,其全部内容通过引用并入本文。
本发明是在国立卫生研究院授予的基金号r01ca109035和r01ca169603的政府支持下完成的。政府对本发明享有一定的权利。
1.技术领域
本发明一般地涉及分子生物学、生物化学、肿瘤学和医学领域。更特别地,本发明涉及用于表征癌细胞的方法和组合物。
2.
背景技术:
大多数癌细胞主要通过胞质溶胶(cytosol)中高速率的糖酵解随后进行乳酸发酵来产生能量(甚至在充足的氧存在下也是如此),而不是像在大多数正常细胞中一样通过线粒体中的丙酮酸氧化产生能量。这种肿瘤特异性瓦博格效应(warburgeffect)促进肿瘤进展(koppenol等,2011;vanderheiden等,2009)。线粒体氧化磷酸化受氧和丙酮酸的可用性的调节,二者分别为该过程的末端电子受体和主要碳源(brown,1992)。线粒体丙酮酸代谢受丙酮酸脱氢酶激酶(pdhk或pdk)(其具有四种同种型(pdhk1-4))和丙酮酸脱氢酶(pdh)的调节(roche和hiromasa,2007)。pdhk1(其表达被缺氧诱导因子1α(hif1α)上调)使pdhe1α亚基的s293磷酸化并使pdh复合物失活,所述pdh复合物通常将丙酮酸转化为乙酰辅酶a(acetyl-coa)和co2;这导致抑制丙酮酸代谢和三羧酸(tca)循环偶联的电子传递,并因此减弱线粒体呼吸和ros产生(holness和sugden,2003;kim等,2006;papandreou等,2006)。通过从线粒体消耗排除丙酮酸,pdhk1诱导可促进糖酵解并提高丙酮酸向乳酸的转化速率。癌细胞还依赖除葡萄糖之外的用于线粒体代谢的多种底物(例如谷氨酰胺)(gao等,2009)。然而,通过癌基因协调调节糖酵解、tca循环和谷氨酰胺分解的机制尚未阐明。
丙酮酸激酶m2(糖酵解途径中的第二atp生成酶)催化磷酸基团从磷酸烯醇丙酮酸(pep)转移至腺苷二磷酸用于产生丙酮酸和腺苷三磷酸(atp)。pkm2还充当使组蛋白h3、bub3、stat3和erk磷酸化的蛋白激酶(yang等,2012,gao等,2012,jiang等,2014;lowery等,2007)。磷酸甘油酸激酶1(pgk1)(糖酵解途径中的第一atp生成酶)催化高能磷酸从1,3-二磷酸甘油酸(1,3-bpg)的1位转移至adp,这导致生成3-磷酸甘油酸(3-pg)和atp(marin-hernandez等,2009;semenza,2010)。在人乳腺癌(zhang等,2005)、胰腺导管腺癌(hwang等,2006)、抗放射性星形细胞瘤(yan等,2012)和多重耐药性卵巢癌细胞(duan等,2002)中以及在转移性胃癌、结肠癌和肝细胞癌细胞(zieker等,2010;ahmad等,2013;ai等,2011)中pgk1表达被上调。尽管其在许多类型的人癌症中过表达,但是pgk1除了其在催化糖酵解反应中的作用之外是否具有其他功能以及pgk1促进的肿瘤发生的机制在很大程度上仍不清楚。
发明概述
如本文中所教导的,缺氧、egfr的活化和k-rasg12v和b-rafv600e的表达诱导erk1/2磷酸化依赖性和pin1顺-反异构化(cis-transisomerization)调节的pgk1线粒体易位。充当蛋白激酶的线粒体pgk1磷酸化并活化pdhk1。不受理论的束缚,这种磷酸化抑制线粒体丙酮酸代谢和ros产生并增强糖酵解和谷氨酰胺分解驱动的脂质合成,从而促进肿瘤发生。
在一个实施方案中,提供了用于治疗患有癌症的患者的组合物,所述癌症被确定包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平。例如,这样的组合物可包含有效量的pgk1抑制剂、mek/erk抑制剂、egfr抑制剂、pin1抑制剂或其组合。在一些方面中,所述组合物可包含至少第二治疗。
在一些方面中,pgk1抑制剂可以是小分子pgk1抑制剂。这样的小分子抑制剂可选择性抑制pgk1的激酶活性。在一些方面中,pgk1抑制剂可包含与pgk1基因的全部或一部分互补的抑制性多核苷酸。这样的抑制性多核苷酸可以是sirna。
在一些方面中,mek/erk抑制剂可以是u0126、azd6244、pd98059、gsk1120212、gdc-0973、rdea119、pd18416、ci1040或fr180204。在一些方面中,egfr抑制剂可以是ag1478。
在一些实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平;以及(b)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。因此,在一个相关实施方案中,提供了包含pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗的组合物用于治疗患有癌症的患者,所述癌症被确定包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的pgk1s203磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的pgk1y324磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的pdhk1t338磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的pdhs293磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的cdc45s386磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的组蛋白h3s10磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的beclin-1s30磷酸化水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在另一个实施方案中,提供了用于治疗患有癌症的患者的方法,其包括(a)选择这样的患者,其癌细胞已被确定包含与参考水平相比升高的线粒体定位的pgk1水平;以及(ii)用pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗和/或pin1抑制剂治疗来治疗所述患者。
在一些实施方案中,提供了选择患有癌症的患者进行pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗的的方法,其包括确定所述患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,其中如果患者包含升高的水平,则选择患者进行pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗。因此,在一些方面中,提供了选择患有癌症的患者进行pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗的方法,其包括:(a)确定患者的癌细胞是否包含升高水平的1、2、3、4、5和/或6中的任一种;以及(b)如果患者的癌细胞包含升高水平的1、2、3、4、5和/或6中的任一种,则选择患者进行pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗。
在一个实施方案中,提供了预测患有癌症的患者中对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗之响应的方法,其包括确定所述患者的癌细胞是否包含:与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高水平的组蛋白h3s10磷酸化;或(7)升高的beclin-1s30磷酸化水平,其中如果来自患者的癌细胞包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,则预测患者对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗具有有利的响应;或者其中如果来自患者的癌细胞不包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,则预测患者不对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗具有有利的响应。在一些方面中,提供了预测患有癌症的患者中对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗之响应的方法,其包括(a)确定患者的癌细胞是否包含与参考水平相比升高水平的1、2、3、4、5和/或6中的任一种;以及(b)如果来自患者的癌细胞包含升高水平的1、2、3、4、5和/或6中的任一种,则将患者鉴定为预测对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗具有有利的响应;或者如果来自患者的癌细胞不包含升高水平的1、2、3、4、5和/或6中的任一种,则将患者鉴定为预测不对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗具有有利的响应。
如在一些实施方案的方法的上下文中使用的,对治疗(例如pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗)的“有利的响应”可包括肿瘤尺寸或负荷减小、肿瘤生长阻滞、肿瘤相关疼痛减轻、癌症相关病理状况减轻、癌症相关症状减轻、癌症无进展、无病间期(diseasefreeinterval)延长、进展时间延长、诱导缓解、转移降低、患者存活延长和/或肿瘤对抗癌治疗的敏感性提高。
在一些方面中,预测响应的方法还包括报告患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化;(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平。在另一些方面中,方法可包括报告癌症是否被预测为对pgk1抑制剂治疗、mek/erk抑制剂治疗、egfr抑制剂治疗或pin1抑制剂治疗有响应。在某些方面中,一些实施方案的方法包括例如通过提供书面、电子或口头报告来报告结果。在一些方面中,向患者提供报告。在另一些方面中,向第三方(例如保险公司或卫生保健提供者(如医生或医院))提供报告。
在一些实施方案中,提供了用于确定患有癌症的患者中预后的方法,其包括确定患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,其中如果癌细胞包含(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,则预测所述患者患有侵袭性癌症(aggressivecancer)。在一些方面中,提供了用于确定患有癌症的患者中预后的方法,其包括确定患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,其中如果癌细胞包含(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平,则预测所述患者患有侵袭性癌症。在一些方面中,提供了用于确定患有癌症的患者中预后的方法,其包括(a)确定患者的癌细胞是否包含与参考水平相比升高水平的1、2、3、4、5、6、7或8中的任一种;以及(b)如果来自患者的癌细胞包含升高水平的1、2、3、4、5、6、7或8中任一种,则将患者鉴定为预测患有侵袭性癌症;或者如果来自患者的癌细胞不包含升高水平的1、2、3、4、5、6、7或8中的任一种,则将患者鉴定为预测不患有侵袭性癌症。
在一些实施方案中,提供了用于确定患有癌症的患者中预后的方法,其包括确定患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化水平或(2)升高的pdhk1t338磷酸化水平,其中如果癌细胞包含(1)升高的pgk1s203磷酸化或y324磷酸化水平或(2)升高的pdhk1t338磷酸化水平,则预测患者患有侵袭性癌症。在一些方面中,提供了用于确定患有癌症的患者中预后的方法,其包括确定患者的癌细胞是否包含与参考水平相比(1)升高的pgk1s203磷酸化或y324磷酸化水平、(2)升高的pdhk1t338磷酸化水平,其中如果癌细胞包含(1)升高的pgk1s203磷酸化或y324磷酸化水平和(2)升高的pdhk1t338磷酸化水平,则预测患者患有侵袭性癌症。因此,在一些方面中,提供了用于确定患有癌症的患者中预后的方法,其包括:(a)确定患者的癌细胞是否包含与参考水平相比升高的pgk1s203磷酸化水平、升高的pgk1y324磷酸化水平、升高的pdhk1t338磷酸化水平、升高的pdhs293磷酸化水平、升高的cdc45s386磷酸化水平、升高的组蛋白h3s10磷酸化水平;以及(b)如果来自患者的癌细胞包含升高的水平,则将患者鉴定为预测患有侵袭性癌症,或者如果来自患者的癌细胞不包含升高的水平,则将患者鉴定为预测不患有侵袭性癌症。
在一些方面中,确定预后的方法可包括确定癌症的等级或癌症将转移的可能性。在某些方面中,确定预后的方法还包括报告来自患者的癌细胞是否包含(1)升高的pgk1s203磷酸化水平、(2)升高的pgk1y324磷酸化水平、(3)升高的pdhk1t338磷酸化水平、(4)升高的pdhs293磷酸化水平、(5)升高的cdc45s386磷酸化水平、(6)升高的组蛋白h3s10磷酸化水平、或(7)升高的beclin-1s30磷酸化水平。在另一些方面中,方法可包括报告癌症是否为侵袭性癌症或报告癌症的等级。在某些方面中,一些实施方案的方法包括例如通过提供书面、电子或口头报告来报告结果。在一些方面,向患者提供报告。在另一些方面中,向第三方(例如保险公司或卫生保健提供者(如医生或医院))提供报告。
在一些方面中,如果预测患者患有侵袭性癌症,则确定预后的方法还可包括向患者施用一种或更多种抗癌治疗。在一些方面中,参考水平可以是来自非癌细胞的水平或来自早期或低度癌细胞的水平。
在另一个实施方案中,提供了用于筛选候选抗癌剂(例如小分子药剂)的方法,其包括在药剂存在或不存在下确定pgk1与pdhk1(或其片段)的结合和/或通过pgk1的pdhk1磷酸化,其中破坏pgk1与pdhk1(或其片段)的结合和/或破坏通过pgk1的pdhk1磷酸化的药剂是候选pgk1抑制剂或抗癌剂。在另一个实施方案中,提供了用于筛选候选pgk1抑制剂或抗癌剂的方法,其包括(a)在药剂存在或不存在下确定pgk1与pdhk1(或其片段)的结合和/或通过pgk1的pdhk1磷酸化;以及(b)基于破坏pgk1与pdhk1(或其片段)的结合和/或破坏通过pgk1的pdhk1磷酸化的药剂,选择候选pgk1抑制剂或抗癌剂。在某些方面中,一些实施方案的用于筛选的方法可涉及筛选小分子、肽和/或多肽(例如,抗体)。在某些方面中,筛选方法可以在无细胞系统中。在另一些方面中,筛选在细胞中(例如,生物体中包含的细胞)进行。在一些情况下,在筛选测定中可包含另外的组分,例如但不限于另外的多肽、脂质、糖类、atp、缓冲剂、螯合剂等。
在一些实施方案中,提供了用于预测患者中癌症严重程度的方法,其包括(a)确定患者样品中pgk1活性的水平、pgk1s203磷酸化的水平、pgk1y324磷酸化的水平或pgk1线粒体定位的水平;以及(b)基于pgk1活性的水平、pgk1s203磷酸化的水平、pgk1y324磷酸化的水平或pgk1线粒体定位的水平,预测对象中癌症严重程度,其中相对于参考水平的升高的pgk1活性、pgk1s203磷酸化或pgk1线粒体定位的水平表示更严重的癌症。在某些方面中,确定pgk1活性的水平可包括确定pdhk1t338磷酸化的水平。
一些实施方案的多个方面涉及确定β-联蛋白(β-catenin)活性的水平;pgk1s203磷酸化的水平;pgk1y324磷酸化的水平;pdhk1t338磷酸化的水平;pdhs293磷酸化的水平;cdc45s386磷酸化的水平;组蛋白h3s10磷酸化的水平;beclin-1s30磷酸化的水平;pgk1线粒体定位的水平;pgk1异构化的水平;和/或pgk1活化的水平。在某些方面中,这种确定可包括进行elisa、免疫测定、放射免疫测定(ria)、免疫组织化学、免疫放射测定、荧光免疫测定、化学发光测定、生物发光测定、凝胶电泳、western印迹分析、southern印迹、流式细胞术、原位杂交、正电子发射断层显像(positronemissiontomography,pet)、单光子发射计算机断层显像(singlephotonemissioncomputedtomography,spect)成像)或显微测定。例如,在一些情况下,使用磷酸化特异性抗体来确定pgk1、pdhk1、pdh、cdc45、组蛋白h3或beclin-1磷酸化的水平。在一些方面中,磷酸化的水平被确定为同一样品中磷酸化蛋白质:未磷酸化蛋白质的比例。在一些方面中,一些实施方案的方法被限定为体外方法,在另一些方面中,方法可在体内进行(例如通过体内成像)。
一些实施方案的一些方面涉及相对于参考水平确定pgk1s203磷酸化的水平;pgk1y324磷酸化的水平;pdhk1t338磷酸化的水平;pdhs293磷酸化的水平;cdc45s386磷酸化的水平;组蛋白h3s10磷酸化的水平;beclin-1s30磷酸化的水平;pgk1线粒体定位的水平;pgk1异构化的水平;和/或pgk1活化的水平。这样的参考水平可以是来自非癌细胞的水平(例如,来自健康患者)或来自早期或低度癌细胞的水平。
一些实施方案的一些方面涉及患者,例如患有癌症的患者。本文中使用的患者可以是人或非人的动物患者(例如,狗、猫、小鼠、马等)。在某些方面中,所述患者患有癌症,例如口腔癌、口咽癌、鼻咽癌、呼吸系统癌症(respiratorycancer)、泌尿生殖系统癌症(urogenitalcancer)、胃肠癌、中枢或周围神经系统组织癌(centralorperipheralnervoussystemtissuecancer)、内分泌或神经内分泌系统癌症(endocrineorneuroendocrinecancer)或造血系统癌症(hematopoieticcancer)、胶质瘤、肉瘤、上皮癌(carcinoma)、淋巴瘤、黑素瘤、纤维瘤、脑脊膜瘤、脑癌、口咽癌、鼻咽癌、肾癌、胆道系统癌症(biliarycancer)、嗜铬细胞瘤、胰岛细胞癌、利-弗劳梅尼瘤(li-fraumenitumor)、甲状腺癌、甲状旁腺癌、垂体瘤、肾上腺瘤、骨源性肉瘤肿瘤(osteogenicsarcomatumor)、神经内分泌系统肿瘤、乳腺癌、肺癌、头颈癌(headandneckcancer)、前列腺癌、食管癌、气管癌、肝癌、膀胱癌、胃癌、胰腺癌、卵巢癌、子宫癌、宫颈癌、睾丸癌、结肠癌、直肠癌或皮肤癌。在一些方面中,癌症为胶质瘤。在一些方面中,癌症为致癌性egfr、致癌性k-ras或致癌性b-raf阳性癌症。
一些实施方案的一些方面涉及患者样品,例如组织样品、流体样品(例如,血液、尿或粪便)或肿瘤活检样品。这样的样品可直接从患者获得或可通过第三方获得。
本文说明书中使用的没有数量词修饰的名词表示一个/种或更多个/种。本文权利要求书中使用的没有数量词修饰的名词,当与词语“包含/包括”结合使用时可表示一个/种或更多个/种。
除非明确地指出仅仅是指替代方案或替代方案是相互排斥的,否则在权利要求书中使用的术语“或/或者”用于意指“和/或”,尽管本公开内容支持仅指替代方案和“和/或”的定义。如本文中使用的“另一个/种”可意指至少第二个/种或更多个/种。
贯穿本申请,术语“约/大约”用于表示这样的值,其包括用于确定所述值所用的设备、方法中误差的固有差异或研究主题之间存在的差异。
从下面的详细描述中,本发明的另一些目的、特征和优点将变得明显。然而,应理解,虽然指示了本发明的优选实施方案,但是详细描述进和具体实施例仅以示例说明的方式给出,因为对本领域技术人员而言,根据该详细描述,本发明的精神和范围内的多种变化和修改将变得明显。
附图简述
以下附图构成本说明书的一部分,并且被包括在内以进一步说明本发明的某些方面。通过结合本文中给出的具体实施方案的详细描述来参考这些附图中的一个或更多个可以更好地理解本发明。
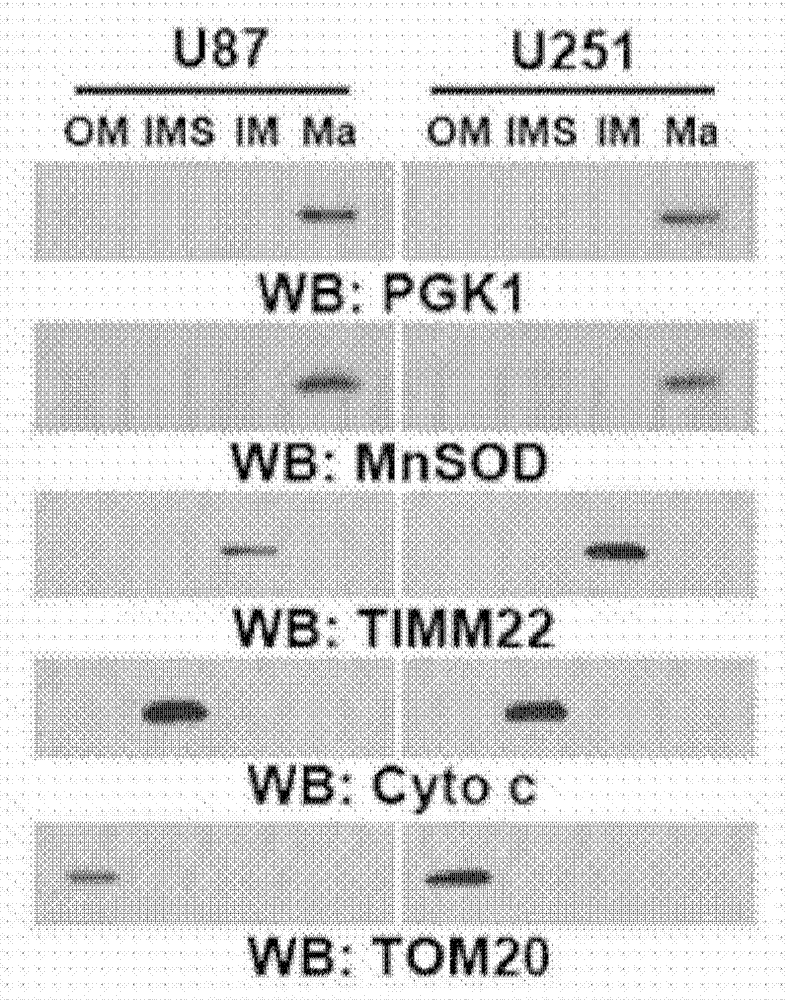
图1a至i.缺氧以及egfr、k-ras和b-raf的活化诱导的pgk1线粒体易位由erk1/2依赖性pgk1s203磷酸化介导。图1b和d至i.使用针对指示的蛋白质的抗体进行免疫印迹和免疫沉淀分析。图1a.将u87细胞缺氧刺激6小时或不进行缺氧刺激并用抗pgk1抗体、mitotracker和dapi进行染色。图1b.将u87和u251细胞缺氧刺激6小时。分离来自线粒体外膜(outermembrane,om)、膜间隙(intermembranespace,ims)、内膜(innermembrane,im)和基质(matrix,ma)的蛋白质。图1c.将u87细胞在缺氧刺激6小时或不进行缺氧刺激。用抗pgk1抗体进行电子显微镜免疫金分析。箭头表示线粒体pgk1的代表性染色。虚线圆表示线粒体。图1d.由u87细胞制备线粒体级分和总细胞裂解物,所述u87细胞用sp600125(25μm)、sb203580(10μm)或u0126(20μm)预处理30分钟,随后缺氧处理6小时。使用微管蛋白作为胞质溶胶蛋白标志物。图1e.将经u0126(20μm)预处理30分钟的egfr过表达的u87(u87/egfr)细胞用egf(100ng/ml)刺激6小时或不用egf刺激。制备线粒体级分和总细胞裂解物。图1f.用表达v5-krasg12v和所指示flag-erk2蛋白的载体稳定地转染bxpc-3细胞或不用其进行转染(左图)。用表达v5-brafv600e和所指示flag-erk2蛋白的载体稳定地转染chl1细胞或不用其进行转染(右图)。制备线粒体级分和总细胞裂解物。图1g.在[γ32p]atp存下,通过将纯化的活性erk2与纯化的wtgst-pgk1或gst-pgk1s203a混合进行体外激酶测定。分离反应混合物用于放射自显影术和免疫印迹分析。图1h.将用表达所指示的sfb标记的pgk1蛋白的载体转染的u87细胞用u0126(20μm)预处理30分钟或不用其进行预处理,随后缺氧刺激6小时。使用链霉亲和素琼脂糖珠拉下sfb标记的蛋白。图1i.将转染有表达所指示的v5标记的pgk1蛋白的载体的u87细胞在缺氧处理6小时或不进行缺氧处理。制备线粒体级分和总细胞裂解物。
图2a至2i.pin1与磷酸化pgk1结合并将其顺-反异构化以用于pgk1线粒体易位。图2a至f和h至i.使用针对所指示蛋白的抗体进行免疫印迹和免疫沉淀分析。图2a.将u87细胞用u0126(20μm)预处理30分钟或不用其进行预处理,随后缺氧刺激6小时。图2b.将u87细胞缺氧刺激处理6小时或不进行缺氧刺激处理。用所指示的gst蛋白进行gst拉下测定(gstpull-downassay)。图2c.将表达所指示pgk1蛋白的u87细胞缺氧刺激处理6小时或不进行缺氧刺激处理。用gst-pin1蛋白进行gst拉下测定。图2d.通过将纯化的重组pgk1与纯化的活性his-erk2混合或不与其混合来进行体外蛋白激酶测定,然后用所指示的gst蛋白进行gst拉下测定测定。图2e.通过将gst-pin1和所指示的纯化的重组pgk1蛋白混合进行gst拉下测定。图2f.将表达所指示的sfb标记的pgk1蛋白的u87细胞缺氧处理6小时或不进行缺氧处理。使用链霉亲和素琼脂糖珠拉下sfb标记的蛋白。图2g.通过将合成的磷酸化或非磷酸化的包含s203p204基序的pgk1的寡肽或包含d203p204基序的pgk1的寡肽与纯化的野生型gst-pin1或gst-pin1c113a突变体混合进行顺-反异构化测定。数据表示三次独立实验的平均值±sd。图2h.重建pin1-/-细胞以表达所指示的pin1蛋白(左图)。由缺氧处理6小时或未进行缺氧处理的所指示细胞制备总细胞裂解物和线粒体级分(右图)。图2i.在pin1+/+细胞和pin1-/-细胞(具有或不具有重建的wtpin1或pin1c113a)中表达v5-pgk1s203d。制备总细胞裂解物和细胞线粒体级分。
图3a至h.pin1调节pgk1与tom复合物的结合。图3a至g.使用针对指示蛋白的抗体进行免疫印迹和免疫沉淀分析。图3a.将u87细胞缺氧处理6小时或不进行缺氧处理。制备总细胞裂解物。图3b.将pin1+/+和所指示的pin1-/-细胞缺氧刺激6小时。图3c.在纯化的his-pin1存在或不存在下,通过将纯化的重组gst-pgk1wt或gst-pgk1s203d与his-tom20混合进行gst拉下测定。图3d至e.将表达所指示sfb标记的pgk1蛋白的u87细胞缺氧处理6小时或不进行缺氧处理。使用链亲和霉素琼脂糖珠拉下sfb标记的蛋白。图3f至g.将表达所指示的v5标记的pgk1蛋白的u87细胞缺氧处理6小时或不进行缺氧处理。制备总细胞裂解物、胞质溶胶级分和线粒体级分。图3h.将表达所指示v5-pgk1蛋白的u87细胞缺氧刺激6小时或不进行缺氧刺激并使用抗v5抗体、mitotracker和dapi进行染色。
图4a至4f.线粒体pgk1使pdhk1磷酸化。图4a至c和e至g.使用所指示的抗体进行免疫印迹分析。图4a.将u87细胞(具有或不具有pgk1shrna且具有或不具有wtrpgk1或rpgk1s203a的重建表达)缺氧刺激6小时或不进行缺氧刺激。制备细胞的线粒体级分,并测量pdh复合物介导的14c标记的丙酮酸向14c标记的co2转化的活性。数据表示三次独立实验的平均值±sd。*p<0.01。图4b.将u87细胞缺氧刺激6小时或不进行缺氧刺激。制备这些细胞的线粒体级分。使用抗pgk1抗体进行免疫沉淀分析。图4c.通过将细菌纯化的sumo-pdhk1蛋白与经纯化的在谷胱甘肽琼脂糖珠上固化的gst或gst-pgk1混合进行gst拉下分析。图4d.在atp存在下通过将细菌纯化的his-pgk1和sumo-pdhk1混合进行体外磷酸化分析。在m/z756.346处的肽的片段谱的质谱结果(质量误差,±4.2ppm)与双电荷肽331-lfnymyp(st)aprpr-343(seqidno:10)匹配,这表明s337或t338被磷酸化。mascot评分为49,期望值:4.7e-004;该匹配的sequest评分为xcorr=3.5。图4e.在[γ32p]atp存在下通过将纯化的wtpgk1或pgk1t378p与纯化的wtpdhk1或pdhk1t338a混合使用放射自显影术进行体外磷酸化分析。图4f.将u251和u87细胞(具有或不具有pgk1shrna且具有或不具有wtrpgk1或rpgk1s203a的重建表达)缺氧刺激6小时或不进行缺氧刺激。
图5a至e.通过pgk1的pdhk1磷酸化使pdhk1活化。使用所指示的抗体进行免疫印迹分析。图5a.将具有纯化的wtpdhk1或pdhk1t338a的细菌纯化的his-pdh与纯化的wtpgk1或pgk1t378p混合。进行体外磷酸化分析。图5b.将表达pgk1shrna的u87和u251细胞(具有或不具有wtrpgk1或rpgk1s203a的重建表达)缺氧刺激6小时或不进行缺氧刺激。图5c.将表达pdhk1shrna的u87和u251细胞(具有或不具有wtrpdhk1或rpdhk1t338a的重建表达)缺氧刺激6小时或不进行缺氧刺激。图5d.将表达pgk1shrna的u87/egfr细胞(具有或不具有wtrpgk1或rpgk1s203a的重建表达)用egf(100ng/ml)刺激6小时或不用egf刺激。图5e.用表达v5-krasg12v和所指示flag-erk2蛋白的载体稳定地转染bxpc-3细胞或不用其进行转染(左图)。用表达v5-brafv600e和所指示flag-erk2蛋白的载体稳定地转染chl1细胞或不用其进行转染(右图)。
图6a至g.pgk1介导的pdhk1磷酸化抑制线粒体丙酮酸代谢、诱导缺氧诱导的ros产生并促进糖酵解。图6a至e.数据表示三次独立实验的平均值±sd。*p<0.01,#p<0.05。图6a.将表达pdhk1shrna的u87细胞(具有或不具有wtrpdhk1或rpdhk1t338a的重建表达)缺氧刺激6小时或不进行缺氧刺激。制备细胞的线粒体级分并测量pdh复合物介导的14c标记的丙酮酸向14c标记的co2转化的活性。图6b.将表达pdhk1shrna的u87细胞(具有或不具有wtrpdhk1或rpdhk1t338a的重建表达)缺氧刺激6小时或不进行缺氧刺激。测量线粒体乙酰辅酶a的水平。图6c.将表达pdhk1shrna的u87细胞(具有或不具有其wt对应物和所指示突变体的重建表达)缺氧刺激24小时或不进行缺氧刺激。测量线粒体ros的水平。图6d.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u87细胞(具有或不具有其wt对应物和所指示突变体的重建表达)缺氧刺激6小时或不进行缺氧刺激。测量胞质溶胶丙酮酸的水平。图6e.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u87细胞(具有或不具有其wt对应物和所指示突变体的重建表达)在缺氧期间在无血清dmem中培养6小时。收集介质用于分析乳酸产生。图6f.将用egf(100ng/ml)刺激24小时或未用egf刺激的u87/egfr细胞用d-[6-14c]-葡萄糖或l-[u-14c]-谷氨酰胺标记2小时。检查细胞的脂质合成。图6g.将具有wtrpgk1或rpgk1s203d之重建表达的内源性pgk1耗竭的u87细胞用l-[u-14c]-谷氨酰胺标记2小时。检查细胞的脂质合成。
图7a至d.线粒体pgk1依赖性pdhk1磷酸化促进细胞增殖和脑肿瘤发生并指示gbm患者中的差的预后。图7a至b.数据表示为三次独立实验的平均值±sd。图7a.在缺氧条件下将总共2×105个u87细胞(具有或不具有pgk1shrna或pdhk1shrna表达且具有或不具有其wt对应物和所指示突变体的重建表达)平板接种(plate)4天。然后收集细胞并计数。图7b.将总共1×106个u87细胞(具有或不具有pgk1shrna或pdhk1shrna表达且具有或不具有其wt对应物和所指示突变体的重建表达)颅内注射到无胸腺的裸小鼠中(每组n=7只小鼠)。在28天后,处死小鼠并检查肿瘤生长。h&e染色的冠状脑切片显示代表性肿瘤异种移植物。使用长a和宽b计算肿瘤体积:v=ab2/2。图7c.对50份人原发性gbm标本用抗磷酸化pgk1s203、抗磷酸化pdhk1t338和抗磷酸化pdhs293抗体进行ihc染色。示出了四个肿瘤的代表性照片。图7d.比较50位患者的存活时间,所述患者具有低(1至4染色评分,顶部曲线)相对于高(4.1至8染色评分,底部曲线)的pgk1s203(低,14位患者;高,36位患者)和pdhk1t338(低,16位患者;高,34位患者)的磷酸化水平。该表显示在调整患者年龄后的多变量分析结果,这表明pgk1s203(p=0.016)和pdhk1t338(p=0.017)磷酸化与患者存活的关联的显著性水平。空心圆表示来自最后一次临床随访时活着的患者的删失数据。
图8.在肿瘤发生中线粒体pgk1协调的糖酵解和tca循环的机制。缺氧或egfr、k-ras和b-raf的活化诱导erk磷酸化和pin1顺-反异构化依赖性pgk1向线粒体中的易位,其中pgk1使pdhk1在t338处磷酸化,导致增强通过pdhk1的pdhs293磷酸化并随后抑制pdh依赖性线粒体丙酮酸代谢和ros产生并提高糖酵解。这种代谢性改变促进细胞增殖和肿瘤发生。虚线箭头:抑制的方向或反应。
图9a至e.缺氧诱导pgk1的线粒体易位。图9a.由缺氧刺激6小时或未进行缺氧刺激的u87和u251细胞制备总细胞裂解物、胞质溶胶和线粒体级分。用所指示的抗体进行免疫印迹分析。缺氧诱导的hif1α表达(左图)是缺氧刺激的对照。wcl:总细胞裂解物;cyto:胞质溶胶;mito:线粒体;wb:western印迹。图9b.由缺氧刺激6小时的u87和u251细胞制备胞质溶胶和线粒体级分。用指示的抗体进行相同百分比的胞质溶胶和线粒体级分的免疫印迹分析。wcl:总细胞裂解物;cyto:胞质溶胶;mito:线粒体。用所指示的抗体进行免疫印迹分析。通过扫描光密度法对图像进行定量。图9c.用hif1αsirna或乱序sirna转染u87细胞,随后缺氧刺激6小时。制备线粒体级分。用所指示的抗体进行免疫印迹分析。图9d.在tritonx-100存在或不存在下,使用蛋白酶k对从缺氧刺激6小时或未进行缺氧刺激的u87或u251细胞分离的线粒体进行处理,然后使用所指示的抗体进行免疫印迹分析。图9e.在毛地黄皂苷(digitonin)(100μm)存在或不存在下,用蛋白酶k对从缺氧刺激6小时或未进行缺氧刺激的u87或u251细胞分离的线粒体进行处理,然后使用所指示的抗体进行免疫印迹分析。timm22(线粒体内膜蛋白)为对照。
图10a至i.pgk1线粒体易位依赖于erk1/2介导的pgk1磷酸化。图10a.从u87细胞制备总细胞裂解物,该u87细胞用sp600125(25μm)、sb203580(10μm)或u0126(20μm)预处理30分钟,随后缺氧处理6小时。用所指示的抗体进行免疫印迹分析。图10b.从用u0126(20μm)预处理30分钟、随后缺氧处理6小时的u251细胞制备线粒体级分。使用所指示的抗体进行免疫印迹分析。图10c.将u87细胞用u0126(20μm)预处理30分钟或不用其进行预处理,随后缺氧处理6小时。使用抗pgk1抗体、mitotracker和dapi进行免疫荧光分析。图10d.将用载体或flag-erk2k52稳定转染的u87细胞缺氧处理6小时或不进行缺氧处理。制备线粒体级分和总细胞裂解物。图10e.用表达具有所指示flag-erk2蛋白的ha-mek1q56p的载体稳定地转染u87细胞或不用其进行转染。制备线粒体级分和总细胞裂解物。使用所指示的抗体进行免疫印迹分析。图10f.将u87细胞缺氧处理6小时或不进行缺氧处理。使用针对指示蛋白的抗体进行免疫印迹和免疫沉淀分析。图10g.用纯化的重组his-erk2孵育固化的gst或gst-pgk1蛋白。用指示的抗体进行免疫印迹分析。图10h.将表达指示的flag标记的erk2蛋白的u87细胞缺氧处理6小时或不进行缺氧处理。使用指示的抗体进行免疫印迹分析。图10i.将转染有表达指示的sfb标记的pgk1蛋白的载体的u87细胞缺氧处理6小时或不进行缺氧处理。使用链霉亲和素琼脂糖珠拉下sfb标记的蛋白质。使用指示的抗体进行免疫印迹分析。
图11a至j.erk1/2介导的pgk1s203磷酸化是pgk1线粒体易位所需的。用指示的抗体进行免疫荧光、免疫沉淀和免疫印迹分析。图11a.将缺氧刺激6小时或未进行缺氧刺激的u87细胞用指示的抗体、mitotracker和dapi进行染色。图11b.将转染有表达指示的sfb标记的pgk1蛋白的载体的u87细胞用u0126(20μm)或sp600125(25μm)预处理30分钟或不用其进行预处理,随后缺氧刺激6小时。使用链霉亲和素琼脂糖珠拉下sfb标记的蛋白。图11c.用表达具有所指示flag-erk2蛋白的ha-mek1q56p的载体稳定地转染u87细胞或不用其进行转染。制备总细胞裂解物。图11d.将表达所指示pgk1蛋白的u87细胞缺氧刺激6小时或不进行缺氧刺激,并使用抗v5抗体、mitotracker和dapi进行染色。图11e.使用抗pgk1ps203抗体耗竭来自总细胞裂解物的磷酸化pgk1,该总细胞裂解物是从缺氧处理6小时或未进行缺氧处理的u87细胞制备的。通过扫描光密度法对图像进行定量。图11f至g.从用ag1478(100nm)或dmso预处理30分钟、随后缺氧处理6小时的u87细胞制备总细胞裂解物。图11h.将稳定表达指示的sfb标记的pgk1蛋白的u87/egfr细胞用egf(100ng/ml)刺激6小时或不用egf刺激。使用链霉亲和素琼脂糖珠拉下sfb标记的蛋白。图11i.将稳定表达指示的v5标记的pgk1蛋白的u87/egfr细胞用egf(100ng/ml)刺激6小时或不用egf刺激。制备线粒体级分和总细胞裂解物。图11j.用表达v5-krasg12v和指示的flag-erk2蛋白的载体稳定地转染bxpc-3细胞或不用其进行转染(上图)。用表达v5-brafv600e和指示的flag-erk2蛋白的载体稳定地转染chl1细胞或不用其进行转染(下图)。
图12a至c.pin1与磷酸化pgk1结合。图12a.使用在线zdock服务器(万维网网址:zdock.umassmed.edu/)进行pgk1(蛋白质数据库id:2zgv)和pin1蛋白(蛋白质数据库id:3tcz)的分子建模分析。pgk1的s203用黄色球表示。pin1的k63和r69用红色带状物表示。图12b.用纯化的野生型gst-pin1孵育固定化的his标记的合成的磷酸化或非磷酸化的包含s203p204基序的pgk1的寡肽或包含d203p204基序的pgk1的寡肽。用指示的抗体进行免疫印迹分析。图12c.从转染有表达v5标记的pgk1s203d蛋白之载体的u87细胞制备胞质溶胶级分和线粒体级分。用指示的抗体对相同百分比的总细胞裂解物、胞质溶胶级分和线粒体级分进行免疫印迹分析。wcl:总细胞裂解物;cyto:胞质溶胶;mito:线粒体。用指示的抗体进行免疫印迹分析。通过扫描光密度法对图像进行定量。
图13a至d.pgk1的前序列中的r39/k41是pgk1线粒体易位所需的。图13a.pgk1的示意性结构示出pgk1的n末端α螺旋中带正电荷的r39、k41和k48。图13b.将表达指示的v5标记的pgk1蛋白的u251细胞缺氧处理6小时或不进行缺氧处理。制备线粒体级分和总细胞裂解物。用指示的抗体进行免疫印迹分析。图13c.pgk1(蛋白质数据库id:2zgv)的结构示出pgk1r39/k41残基(黄色球)未暴露在pgk1蛋白的表面上。图13d.将纯化的gst-pgk1s203d与纯化的pin1混合或不与其混合,然后用识别38-qrikaa-43的特异性抗pgk1抗体进行免疫沉淀。用抗gst抗体进行免疫印迹分析。
图14a至p.线粒体pgk1使pdhk1在t338处磷酸化。图14a.将u87细胞(具有或不具有pgk1shrna且具有或不具有wtrpgk1或rpgk1r39/k41a的重建表达)缺氧刺激6小时或不进行缺氧刺激。制备细胞的线粒体级分并测量pdh复合物介导的14c标记的丙酮酸向14c标记的co2转化的活性。数据表示三次独立实验的平均值±sd。*p<0.01。用指示的抗体进行免疫印迹分析。图14b.将表达pgk1shrna的u87细胞(左图)或u87/egfr细胞(右图)(具有或不具有wtrpgk1和所指示突变体的重建表达)缺氧刺激6小时或不进行缺氧刺激(左图),或者用egf(100ng/ml)刺激6小时或不用egf进行刺激(右图)。将这些细胞用[1-14c]-丙酮酸孵育2小时。测量[1-14c]co2产生速率。图14c.将表达sfb-pdhk1、sfb-pdhk2、sfb-pdhk3或sfb-pdhk4的u87细胞缺氧刺激6小时或不进行缺氧刺激。制备这些细胞的线粒体级分。用链霉亲和素琼脂糖珠进行sfb拉下分析。用指示的抗体进行免疫印迹分析。图14d.通过在[γ32p]atp存在下在激酶测定条件下将细菌纯化的his-pgk1和sumo-pdhk1混合进行体外磷酸化分析。进行凝胶分离的32p磷酸化的sumo-pdhk1的磷酸化-氨基酸分析。左侧图示出了经染色的磷酸化-氨基酸标志物。在右侧图的放射自显影图上,绿色圆表示pser;红色圆表示pthr;黑色圆表示ptyr。图14e.在[γ32p]atp存在下通过将纯化的wtpgk1或pgk1t378p与纯化的wtpdhk1、pdhk1s337a或pdhk1t338a混合用放射自显影术进行体外磷酸化分析。图14f.通过在3-pg(0.5mm)存在或不存在下将纯化的pgk1与纯化的pdhk1混合进行体外磷酸化分析。使用指示的抗体进行免疫印迹分析。图14g.通过在甘油醛3-磷酸、nad+和甘油醛磷酸脱氢酶(gapdh)存在下将纯化的wtpgk1或pgk1t378p与纯化的pdhk1混合进行体外磷酸化分析。用指示的抗体进行免疫印迹分析。图14h.将从u87细胞制备的线粒体级分用atp酶(1单元/mg裂解物蛋白质)处理或不用其进行处理,随后用纯化的wtpgk1孵育。用指示的抗体进行免疫印迹分析。图14i.pgk1的km和1/v相对于1/[atp]的代表性图。图14j.测量从缺氧处理6小时或未进行缺氧处理的u87或u251细胞分离的线粒内膜体(mitoplast)中的atp浓度。图14k.用抗pdhk1pt338抗体免疫耗竭缺氧处理6小时或未进行缺氧处理的u87细胞的线粒体级分或者不用所述抗体进行免疫耗竭。用指示的抗体进行免疫印迹分析。图14l.重建表达pgk1shrna的u87和u251细胞以表达wtrpgk1或rpgk1t378p。用指示的抗体进行免疫印迹分析。图14m至n.将u87和u251细胞(具有pgk1shrna且具有wtrpgk1或rpgk1t378p的重建表达)的缺氧刺激6小时或不进行缺氧刺激。制备线粒体级分。用指示的抗体进行免疫印迹分析。图14o.测量细菌纯化的wtpgk1、pgk1t378p、pgk1s203a和pgk1r39/k41a的糖酵解活性。数据表示三次独立实验的平均值±sd。用抗pgk1抗体进行免疫印迹分析。图14p.将u87细胞缺氧刺激6小时或不进行缺氧刺激。用指示的抗体进行线粒体裂解物的免疫印迹分析。
图15a至c.线粒体pgk1以pdhk1t338磷酸化依赖性方式导致增强的pdh磷酸化。图15a.将表达pgk1shrna的u87和u251细胞(具有或不具有wtrpgk1或rpgk1t378p的重建表达)缺氧刺激6小时或不进行缺氧刺激。使用指示的抗体进行线粒体裂解物的免疫印迹分析。图15b.将表达pgk1shrna的u87(具有或不具有wtrpgk1或rpgk1r39/k41a的重建表达)缺氧刺激6小时或不进行缺氧刺激。用指示的抗体进行线粒体裂解物的免疫印迹分析。图15c.将表达pgk1shrna的u87(具有wtrpgk1或rpgk1r39/k41a的重建表达)缺氧刺激24小时或不进行缺氧刺激。用指示的抗体进行线粒体裂解物的免疫印迹分析。
图16a至m.pgk1介导的pdhk1磷酸化抑制线粒体丙酮酸代谢并促进糖酵解。图16a至k.数据表示三次独立实验的平均值±sd。*p<0.01。图16a.将表达pgk1shrna的u87细胞(左图)或u87/egfr细胞(右图)(具有或不具有wtrpgk1和所指示突变体的重建表达)缺氧刺激6小时或不进行缺氧刺激(左图),或者用egf(100ng/ml)刺激6小时或不用egf进行刺激(右图)。将这些细胞用[1-14c]-丙酮酸孵育2小时。测量[1-14c]co2产生速率。图16b.将表达pdhk1shrna的u251细胞(具有或不具有wtrpdhk1或rpdhk1t338a的重建表达)缺氧刺激6小时或不进行缺氧刺激。测量线粒体乙酰辅酶a的水平。图16c.将表达pdhk1shrna的u251细胞(具有或不具有wtrpdhk1或rpdhk1t338a的重建表达)缺氧刺激24小时或不进行缺氧刺激。测量线粒体ros的水平。图16d.用不同表达水平的wtrpdhk1或rpdhk1t338a重建内源性pdhk1耗竭的u87细胞(左图),并缺氧刺激24小时。在缺氧条件下测量细胞的线粒体ros产生(右图)。l:低表达;h:高表达。用指示的抗体进行免疫印迹分析。图16e.将内源性pgk1耗竭的u87细胞(具有wtrpgk1或rpgk1r39/k41a的重建表达)缺氧刺激24小时或不进行缺氧刺激。测量线粒体ros的水平。图16f.将具有或不具有pgk1耗竭的u87细胞缺氧刺激6小时。向分离的线粒体中添加指定浓度的丙酮酸。测量线粒体ros的水平。图16g.将具有或不具有pgk1耗竭的u87细胞缺氧刺激6小时。向分离的线粒体中添加指定浓度的丙酮酸。测量线粒体膜电位的水平。aψm:线粒体膜电位。图16h.将内源性pgk1耗竭的u87/egfr细胞(具有wtrpgk1或rpgk1s203a的重建表达)用dca(5mm)预处理1小时,随后用egf(100ng/ml)刺激6小时。测量线粒体ocr。图16i.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u251细胞(具有或不具有其wt对应物和所指示突变体的重建表达)缺氧刺激6小时或不进行缺氧刺激。测量胞质溶胶丙酮酸的水平。图16j.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u251细胞(具有或或不具有其wt对应物和所指示突变体的重建表达)在缺氧期间在无血清dmem中培养6小时。收集介质用于分析乳酸产生。图16k.将表达pgk1shrna的u251细胞(具有或或不具有wtrpgk1或rpgk1s203d的重建表达)在正常氧条件下在无血清dmem中培养6小时。收集介质用于分析乳酸产生。图16l.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u87/egfr细胞(具有或或不具有其wt对应物和所指示突变体的重建表达)用egf(100ng/ml)刺激6小时或不用egf刺激。制备线粒体级分并测量pdh复合物介导的14c标记的丙酮酸向14c标记的co2转化的活性。用指示的抗体进行免疫印迹分析。图16m.将表达pgk1shrna(左图)或pdhk1shrna(右图)的u87/egfr(具有或不具有其wt对应物和所指示突变体的重建表达)在egf(100ng/ml)刺激期间在无血清dmem中培养6小时。收集介质用于分析乳酸产生。
图17a至h.线粒体pgk1依赖性pdhk1磷酸化促进细胞增殖和脑肿瘤发生。图17a至c.数据表示三次独立实验的平均值±sd。图17a.将总共2×105个u251细胞(具有或不具有pgk1shrna(左图)或pdhk1shrna(右图)表达且具有或不具有其wt对应物和所指示突变体的重建表达)在缺氧条件下平板接种4天。然后收集细胞并计数。图17b.将具有wtrpgk1或rpgk1r39/k41a的重建表达的总共2×105个内源性pgk1耗竭的u87细胞平板接种,并在缺氧期间用dtt(1mm)处理4天或不用dtt处理。然后收集细胞并计数。图17c.制备来自具有wtrpgk1或rpgk1s203a的重建表达的内源性pgk1耗竭的u87/egfrviii细胞的线粒体级分和总细胞裂解物。用指示的抗体进行免疫印迹分析(左图)。将指示的u87/egfrviii细胞(2×105)平板接种3天并计数(右图)。图17d.将具有wtrpgk1或rpgk1s203d的重建表达的总共2×105个内源性pgk1耗竭的u87细胞平板接种并用谷氨酰胺饥饿处理3天或不用其进行饥饿处理。然后收集细胞并计数。#p<0.05。图17e.具有pgk1shrna(上图)或pdhk1shrna(下图)表达的gsc11细胞重建表达其wt对应物和所指示突变体。用指示的抗体进行总细胞裂解物的免疫印迹分析。图17f.将总共1×106个gsc11细胞(具有或不具有pgk1shrna(上图)或pdhk1shrna(下图)表达且具有或不具有其wt对应物和所指示突变体的重建表达)颅内注射到每组的无胸腺裸小鼠中。在21天后,处死小鼠并检查肿瘤生长。h&e染色的冠状脑切片示出了代表性肿瘤异种移植物。采用长a和宽b计算肿瘤体积:v=ab2/2。图17g.使用抗ki67抗体进行肿瘤组织的ihc分析。在10个显微镜视野中定量ki67阳性细胞。图17h.进行指示的肿瘤组织的tunel分析。将凋亡的细胞染成褐色。在10个显微镜视野中对凋亡的细胞进行定量。
图18.erk介导的pgk1磷酸化和pgk1依赖性pdhk1磷酸化与pdh磷酸相关联。用抗磷酸化pgk1s203、抗磷酸化pdhk1t338和抗磷酸化pdhs293抗体对50份人原发性gbm标本进行ihc染色。进行半定量评分(级别,1至8)(皮尔逊积差相关检验;左上图,r=0.66,p<0.01;右上图,r=0.71,p<0.01;下图,r=0.73,p<0.01)。注意图上的一些点表示多于一个标本(即,一些评分重叠)。
图19a至c.pgk1使组蛋白h3、cdc45和beclin-1磷酸化。图19a.组蛋白h3ps10的western印迹示出pgk1使组蛋白h3在s10处磷酸化。在对照条件下,egf刺激组蛋白h3在s10处的磷酸化,然而,这种作用通过使用shrna敲减pgk1而大幅减弱。图19b.cdc45的western印迹和放射自显影术印迹示出pgk1使cdc45在ps386处磷酸化。在[γ32p]-atp存在下,纯化的野生型pgk1而非pgk1激酶失活(kinase-dead,kd)突变体使纯化的野生型cdc45而非cdc45s386a磷酸化。图19c.beclin-1的western印迹和放射自显影术印迹示出pgk1使beclin-1在s30处磷酸化。在[γ32p]-atp存在下,纯化的pgk1使野生型beclin-1磷酸化。beclin-1s30a突变体在很大程度上抵抗通过pgk1的磷酸化。
图20a至c.图20a至b.通过将纯化的重组pgk1wt、y324f或t378p与[γ32p]-atp混合进行体外蛋白激酶测定。通过放射自显影术(图20a)或pgk1py324抗体(图20b)检测pgk1y324磷酸化。图20c.pgk1酶活性测定。将纯化的重组pgk1wt、y324f或t378p在25℃下在96孔板中在100μl反应缓冲液中(50mmtris-hcl[ph7.5]、5mmmgcl2、5mmatp、0.2mmnadh、10mm甘油-3-磷酸酯和10u的gapdh)孵育并在动力学模式下在339nm处读取5分钟。
具体实施方式
瓦博格效应的特征在于与抑制的线粒体丙酮酸代谢组合的提高的葡萄糖摄取和乳酸产生。然而,胞质溶胶糖酵解与线粒体代谢协调调节的机制尚未阐明。缺氧、egfr活化以及k-rasg12v和b-rafv600e的表达诱导磷酸甘油酸激酶1(pgk1)的线粒体易位;这是通过pgk1在s203处的erk依赖性磷酸化和随后的ps203.p204的pin1介导的顺-反异构化介导的,允许pgk1n末端α螺旋中的r39/k41与tom复合物相互作用。在线粒体中,pgk1充当蛋白激酶来使丙酮酸脱氢酶激酶1(pdhk1)在t338处磷酸化,这使pdhk1活化以磷酸化并抑制丙酮酸脱氢酶(pdh)复合物。这种pgk1介导的pdh抑制降低丙酮酸在线粒体中的利用、抑制活性氧类产生并且提高糖酵解和谷氨酰胺分解驱动的脂质合成,导致提高的肿瘤发生。此外,pgk1s203和pdhk1t338磷酸化水平与pdhs293磷酸化水平和胶质母细胞瘤患者的差的预后相关联。这些发现揭露了pgk1作为蛋白激酶在协调糖酵解和tca循环中的功能,这有助于癌症代谢和肿瘤发生。
本发明说明了pgk1是包含可以靶向用于癌症治疗的蛋白激酶活性的代谢酶。pgk1是用于肿瘤发生的蛋白激酶;磷酸化事件pgk1ps203、pdhkpt338、cdc45ps386、beclin1ps30和组蛋白h3ps10是用于预后和个性化治疗的生物标志物。
i.本发明的一些方面
由于远离脉管系统的大量扩增,最初在血管环境中发生的肿瘤细胞团可变得严重缺氧(brahimi-hom等,2007)。为了在这种缺氧应激下存活以及为了支持肿瘤细胞生长,肿瘤细胞上调糖酵解并抑制线粒体中的丙酮酸代谢和氧化磷酸化,使得丙酮酸在胞质溶胶中转化为乳酸(gatenby和gillies,2004)。在正常氧条件下,受体酪氨酸激酶的活化或存在普遍的k-ras和b-raf突变促进瓦博格效应(yang等,2012;yaffe等,1997;tani等,1985)。不受理论的束缚,本文中证明的是先前未知的通过糖酵解酶pgk1的亚细胞区室依赖性调节之协调调节糖酵解和tca循环的机制:缺氧应激、egfr的活化或者k-rasg12v或b-rafv600e的表达导致pgk1在s203处erk1/2依赖性磷酸化,导致pin1依赖性pgk1顺-反异构化、pgk1与tom复合物的结合以及随后的pgk1线粒体易位。在线粒体中,pgk1直接与pdhk1在t338处相互作用并使其磷酸化。这种磷酸化导致增强的pdhk1活性和pdhk1介导的pdh磷酸化,这导致抑制线粒体中的pdh依赖性丙酮酸利用和ros产生,并提高糖酵解以及egf诱导的和谷氨酰胺分解促进的脂质合成。这种代谢性改变促进了细胞增殖和肿瘤发生(图8)。连同pkm2发挥组蛋白激酶功能作用的先前发现(yang等,2012),pgk1发挥蛋白激酶功能作用的证明突出了pgk1和pkm2的双重作用,这在细胞代谢和增殖中充当糖酵解酶和蛋白激酶二者,扩展了重要家族分支的激酶组(kinome),并且极大地影响了对具有“多面”的蛋白酶在控制细胞功能方面的理解。
缺氧细胞提高糖酵解以及抑制细胞呼吸。提高的糖酵解主要归因于hif1α-或hif2α-依赖性糖酵解酶表达,而抑制细胞呼吸被认为是由于接受来自线粒体呼吸链的电子所需的氧的缺乏以及由于线粒体丙酮酸代谢和呼吸的抑制,这可受多种机制(包括hif1α上调的pdhk1表达)调节(kim等,2006;semenza,2008)。通过表达pgk1r39/k41a诱导的pgk1线粒体易位的缺陷(其对缺氧增强的pdhk1表达没有影响)很大程度上降低缺氧诱导的pdh磷酸化。这些结果表明pdhk1通过自身的过表达不足以使其在线粒体中的细胞活性最大化,并且pgk1介导的pdhk1的磷酸化和缺氧增强的pdhk1表达对调节pdhk1活性具有协同作用。
尽管肿瘤细胞可在缺氧条件下通过hif调节的糖酵解基因表达和线粒体酶同时调节糖酵解和线粒体,但是这种调节(其在正常氧条件下不发生)是慢性反应并且需要调节基因转录。在正常氧条件下egfr、k-ras和b-raf的活化或缺氧刺激诱导肿瘤细胞通过pgk1的迅速线粒体易位的立即或急性响应,这导致抑制线粒体丙酮酸代谢、线粒体丙酮酸穿梭至胞质溶胶用于乳酸产生、以及提高谷氨酰胺分解促进的脂肪酸合成。因此,这些发现提供了糖酵解、tca循环和谷氨酰胺分解的综合调节的新概念,并且提供了对于由普遍存在的癌基因(例如egfr、k-ras和b-raf)诱导的瓦博格效应之关键的新见解。鉴于pgk1表达在人癌症中上调并且与肿瘤转移和耐药性相关(zhang等,2005;hwang等,2006;duan等,2002;zieker等,2010;ahmad等,2013;ai等,2011),这些证明了pgk1依赖性pdhk1t338磷酸化促进肿瘤细胞增殖和肿瘤发生以及pgk1s203和pdhk1t338磷酸化水平与胶质母细胞瘤预后相关联的发现提供了用于改进诊断和治疗人癌症的分子基础。
ii.检测方法
在某些实施方案中,所述方法包括以下步骤:从待测试的哺乳动物获得生物样品;检测样品中pgk1、pdhk1、pdh、cdc45、组蛋白h3或beclin-1蛋白的磷酸化水平。在一个实施方案中,生物样品是来自哺乳动物中肿瘤的细胞样品。如本文中使用的短语“选择性测量”是指其中仅在样品中测量有限数量的蛋白质磷酸化事件而非测定基本上所有蛋白质磷酸化的方法。例如,在一些方面中,“选择性测量”蛋白质磷酸化事件可指测量不多于100、75、50、25、15、10、5或2个不同的蛋白质磷酸化事件。
在本文中描述的方法的另一个实施方案中,检测从个体获得的生物样品中磷酸化蛋白质的存在包括确定样品中磷酸化多肽的水平。磷酸化蛋白质的水平可通过使样品与特异性结合磷酸化多肽的抗体接触并确定结合的抗体的量来确定(例如通过检测或测量抗体与多肽之间复合物的形成)。抗体可被标记(例如,放射性、荧光、生物素化或hrp缀合)以便于检测复合物。用于检测多肽水平的合适的测定系统包括但不限于:流式细胞术、酶联免疫吸附测定(elisa)、竞争elisa测定、放射免疫测定(ria)、免疫荧光、凝胶电泳、western印迹和化学发光测定、生物发光测定、涉及使用对多肽产物具有特异性的抗体在样品中测定磷酸化蛋白质的免疫组织化学测定。用于本发明的检测和分析的多种方法和设备是技术人员熟知的。关于受试样品中的多肽或蛋白质,经常使用免疫测定设备和方法。这些设备和方法可以以多种夹心、竞争性或非竞争性测定方式利用标记分子以产生与目的分析物的存在或量有关的信号。另外地,可以使用某些方法和设备(例如但不限于生物传感器和光学免疫测定)在无需标记的分子的情况下确定分析物的存在或量。
或者,可使用质谱分析检测pgk1、pdhk1、pdh、cdc45、组蛋白h3或beclin-1多肽的磷酸化水平。已经使用质谱分析来检测血清样品中的蛋白质。质谱方法包括表面增强激光解吸电离(surfaceenhancedlaserdesorptionionization,seldi)质谱(ms)、seldi飞行时间质谱(tof-ms)、maldiqqtof、ms/ms、tof-tof、esi-q-tof和ion-trap。
可通过本领域技术人员已知的多种方式中任一种对多肽进行检测和定量,包括分析生物化学方法,例如电泳、毛细管电泳、高效液相色谱(“hplc”)、薄层色谱(“tlc”)、高扩散色谱等;或多种免疫学方法,例如流体或凝胶沉淀反应、免疫扩散(单向或双向)、免疫电泳、放射免疫测定(“ria”)、酶联免疫吸附测定(“elisa”)、免疫荧光测定、流式细胞术、facs、western印迹等。
还可使用免疫组织化学染色来测量多种生物标志物的差异表达。这种方法能够通过蛋白质与特异性抗体的相互作用来定位组织切片的细胞中的蛋白质。为此,可将组织固定在甲醛或其他合适的固定剂中,包埋在石蜡或塑料中并使用切片机将其切成薄切片(约0.1mm至数mm厚)。或者,可使用低温恒温器将组织冷冻并切成薄切片。可将组织的切片排列在固体表面上并附着于其上(即,组织微阵列)。将组织切片与针对目的抗原的一抗一起孵育,然后洗涤以除去未结合的抗体。可将一抗与检测系统偶联,或可用与检测系统偶联的二抗检测一抗。检测系统可以是荧光团或其可以是酶(例如辣根过氧化物酶或碱性磷酸酶),其可将底物转化成比色、荧光或化学发光产物。通常在显微镜下扫描经染色的组织切片。因为来自患有癌症的对象的组织样品可以是异质的,即一些细胞可以是正常并且另一些细胞可以是癌性的,所以可确定组织中阳性染色细胞的百分比。可以使用这种测量与染色强度定量一起来生成生物标志物的表示值。
可以使用酶联免疫吸附测定或elisa来测量多种生物标志物的差异表达。存在多种变化形式的elisa测定。所有都基于抗原或抗体在固体表面(通常是微量滴定板)上的固定化。最初的elisa方法包括制备包含目的生物标志物蛋白的样品,用样品包被微量滴定板的孔,用识别特异性抗原的一抗孵育每个孔,洗掉未结合的抗体,然后检测抗体-抗原复合物。可直接检测抗体-抗原复合物。为此,将一抗与检测系统(例如产生可检测产物的酶)偶联。可间接检测抗体-抗原复合物。为此,如上所述通过与检测系统偶联的二抗检测一抗。然后扫描微量滴定板并且可使用本领域已知的方法将原始强度数据转化成表示值。
还可使用抗体微阵列来测量多种生物标志物的差异表达。为此,将多种抗体排列并共价地附着至微阵列或生物芯片的表面。通常用荧光染料标记包含目的生物标志物蛋白的蛋白质提取物。将标记的生物标志物蛋白与抗体微阵列一起孵育。在洗涤以除去未结合的蛋白质后,扫描微阵列。可以使用本领域已知的方式将原始荧光强度据转化成表示值。
iii.治疗方法
本发明的某些方面可用于基于pgk1的s203、pgk1的y324、pdhk1的t338、pdh的s293、cdc45的s386、组蛋白h3的s10和/或beclin-1的s30的磷酸化状态用来鉴定和/或治疗疾病或病症。本发明的另一些方面提供了用pgk1、mek/erk、egfr和/或pin1抑制剂治疗癌症患者。
本文中使用的术语“对象”或“患者”是指对其执行主题方法的任何个体。尽管如本领域技术人员将理解的,患者通常是人,但是患者可以是动物。因此,在患者的定义中包括其他动物,包括哺乳动物,例如啮齿动物(包括小鼠、大鼠、仓鼠和豚鼠)、猫、狗、兔、农场动物(包括牛、马、山羊、绵羊、猪等)和灵长类(包括猴、黑猩猩、猩猩和大猩猩)。
“治疗”是指为了获得疾病或健康相关病症的治疗益处而向对象施用或施加治疗剂或对对象执行某种方法或方式。例如,治疗可包括施用化学治疗、免疫治疗、放射治疗、进行手术或其任意组合。
贯穿本申请使用的术语“治疗益处”或“治疗有效的”是指就药物治疗该病症而言促进或增强对象健康的任何情况。这包括但不限于疾病的体征或症状的频率或严重程度降低。例如,癌症的治疗可涉及例如肿瘤侵袭性降低、癌症生长速率降低或阻止转移。癌症的治疗还可指患癌症对象存活的延长。
包括组合治疗的方法和组合物增强治疗或预防效果,和/或提高其他抗癌或抗过度增殖治疗的治疗效果。可以以有效实现期望效果的组合量提供治疗和预防的方法和组合物。可以使组织、肿瘤或细胞与包含一种或更多种药剂的一种或更多种组合物或药理学制剂接触,或者通过使组织、肿瘤和/或细胞与两种或更多种不同的组合物或制剂接触。此外,考虑这样的组合治疗可与化学治疗、放射治疗、手术治疗或免疫治疗联合使用。
当应用于细胞时,在本文中使用的术语“接触”或“暴露”用于描述通过其治疗构建体和化学治疗剂或放射治疗剂被递送至靶细胞或被置于与靶细胞的直接临近处的方法。为了实现细胞杀伤,例如这两种药剂以有效杀伤细胞或阻止其分裂的组合量递送至细胞。
本文中描述的方法可用于治疗癌症。通常来说,术语“癌症”或“癌性的”是指或描述哺乳动物中以细胞生长失控为典型特征的生理学状态。更具体地,使用任意一种或更多种pgk1、mek/erk、egfr和/或pin1抑制剂或其变体并结合本文中提供的方法治疗的癌症包括但不限于实体肿瘤、转移癌或非转移癌。在某些实施方案中,癌症可以起源于膀胱、血液、骨、骨髓、脑、乳腺、结肠、食管、十二指肠、小肠、大肠、结肠、直肠、肛门、牙龈(gum)、头、肾、肝、肺、鼻咽、颈、卵巢、胰腺、前列腺、皮肤、胃、睾丸、舌或子宫。
癌症可以具体地具有以下组织学类型,但其不限于这些:恶性赘生物;上皮癌;淋巴瘤;母细胞瘤;肉瘤;未分化的上皮癌;脑脊膜瘤;脑癌;口咽癌;鼻咽癌;肾癌;胆道系统癌症;嗜铬细胞瘤;胰岛细胞癌;利-弗劳梅尼瘤;甲状腺癌;甲状旁腺癌;垂体瘤;肾上腺瘤;骨源性肉瘤肿瘤;神经内分泌系统肿瘤;乳腺癌;肺癌;头颈癌;前列腺癌;食管癌;气管癌;肝癌;膀胱癌;胃癌;胰腺癌;卵巢癌;子宫癌;宫颈癌;睾丸癌;结肠癌;直肠癌;皮肤癌;巨细胞和梭状细胞癌;小细胞癌;小细胞肺癌;非小细胞肺癌;乳头状癌;口腔癌;口咽癌;鼻咽癌;呼吸系统癌症;泌尿生殖系统癌症;鳞状细胞癌;淋巴上皮癌;基底细胞癌;毛母质癌;移行细胞癌;乳头状移行细胞癌;腺癌;胃肠癌;恶性胃泌素瘤;胆管癌;肝细胞癌;联合肝细胞癌和胆管癌;小梁腺癌;腺样囊性癌;腺瘤性息肉中的腺瘤;腺癌,家族性结肠息肉病;实体癌;恶性类癌肿瘤;分支肺泡腺癌;乳头状腺癌;嫌色细胞癌;嗜酸性癌;嗜酸性腺癌;嗜碱性癌;透明细胞腺癌;颗粒细胞癌;滤泡腺癌;乳头状腺癌和滤泡状腺癌;非包囊硬化性癌(nonencapsulatingsclerosingcarcinoma);肾上腺皮质癌;子宫内膜癌;皮肤附属器癌;大汗腺癌(apocrineadenocarcinoma);皮脂腺癌;耵聍腺癌(ceruminousadenocarcinoma);黏液表皮样癌;囊腺癌;乳头状囊腺癌;乳头状浆液性囊腺癌(papillaryserouscystadenocarcinoma);黏液性囊腺癌;黏液性腺癌;印戒细胞癌;浸润性导管癌;髓质癌;小叶癌;炎性癌;乳腺佩吉特癌;腺泡细胞癌;腺鳞癌;腺癌伴鳞状化生(adenocarcinomawithsquamousmetaplasia);恶性胸腺瘤;恶性卵巢基质瘤;恶性卵泡膜细胞瘤(thecoma);恶性颗粒细胞瘤;恶性睾丸母细胞瘤(androblastoma);赛托利细胞癌(sertolicellcarcinoma);恶性莱迪希细胞瘤(leydigcelltumor);恶性脂质细胞瘤;恶性副神经节瘤;恶性乳腺外副神经节瘤;嗜铬细胞瘤;肾小球肉瘤(glomangiosarcoma);恶性黑素瘤;无色素性黑素瘤;浅表扩散性黑素瘤;巨型色素痣中的恶性黑素瘤;恶性雀斑样痣黑素瘤(lentigomalignamelanoma);肢端雀斑样痣黑素瘤(acrallentiginousmelanoma);结节性黑素瘤;上皮样细胞黑素瘤;恶性蓝色痣;肉瘤;纤维肉瘤;恶性纤维组织细胞瘤;黏液肉瘤;脂肪肉瘤;平滑肌肉瘤;横纹肌肉瘤;胚胎性横纹肌肉瘤;肺泡横纹肌肉瘤;基质肉瘤;恶性混合瘤;苗勒氏混合肿瘤(mullerianmixedtumor);肾母细胞瘤;肝母细胞瘤;癌肉瘤;恶性间质瘤;恶性布伦纳瘤;恶性叶状瘤(phyllodestumor);滑膜肉瘤;恶性间皮瘤;无性细胞瘤;胚胎癌;恶性畸胎瘤;恶性卵巢甲状腺瘤(strumaovarii);绒毛膜癌;恶性中肾瘤;血管肉瘤;恶性血管内皮细胞瘤;卡波西肉瘤;恶性血管外皮细胞瘤;淋巴管肉瘤;骨肉瘤;近皮质骨肉瘤(juxtacorticalosteosarcoma);软骨肉瘤;恶性软骨母细胞瘤;间充质软骨肉瘤(mesenchymalchondrosarcoma);骨巨细胞瘤;尤因肉瘤;恶性牙肉瘤(odontosarcoma);成釉细胞牙肉瘤;恶性成釉细胞瘤;成釉细胞纤维肉瘤;内分泌或神经内分泌系统癌症或造血系统癌症;恶性松果体瘤;脊索瘤;中枢或周围神经系统组织癌;恶性胶质瘤;室管膜瘤;星形细胞瘤;原生质型星形细胞瘤;纤维性星形细胞瘤;星形母细胞瘤;胶质母细胞瘤;少突神经胶质肿瘤;少突胶质母细胞瘤;原发性神经外胚层瘤;小脑肉瘤;神经节神经母细胞瘤;神经母细胞瘤;视网膜母细胞瘤;嗅神经源性肿瘤;恶性脑脊膜瘤;神经纤维肉瘤;恶性神经鞘瘤;恶性颗粒细胞瘤;b细胞淋巴瘤;恶性淋巴瘤;霍奇金病;霍奇金病;低度/滤泡性非霍奇金淋巴瘤;副肉芽肿;小淋巴细胞恶性淋巴瘤;大细胞弥漫性恶性淋巴瘤;滤泡性恶性淋巴瘤;蕈样肉芽肿(mycosisfungoides);套细胞淋巴瘤;瓦尔登斯特伦巨球蛋白血症(waldenstrom’smacroglobulinemia);其他特定的非霍奇金淋巴瘤;恶性组织细胞增生症;多发性骨髓瘤;肥大细胞肉瘤;免疫增生性小肠疾病;白血病;淋巴性白血病;浆细胞白血病;红白血病;淋巴肉瘤细胞白血病;髓性白血病;嗜碱性白血病;嗜酸粒细胞白血病;单核细胞白血病;肥大细胞白血病;巨核细胞白血病;骨髓肉瘤;慢性淋巴细胞白血病(cll);急性淋巴细胞白血病(all);毛细胞白血病;慢性粒细胞白血病;和毛细胞白血病。
患者对治疗的有效响应或患者对治疗的“响应性”是指给予处于疾病或病症风险或遭受疾病或病症的患者的临床或治疗益处。这样的益处可以包括细胞或生物学响应、完全响应、部分响应、稳定疾病(没有进展或复发)或具有较晚复发的响应。例如,在诊断患有癌症的患者中有效响应可以是减小的肿瘤尺寸或无进展存活。
关于肿瘤病症治疗,根据肿瘤病症的阶段,肿瘤病症治疗涉及以下治疗中的一种或组合:除去肿瘤组织的手术、放射治疗和化学治疗。其他治疗方案可以与施用抗癌剂(例如,治疗组合物和化学治疗剂)组合。例如,待用这样的抗癌剂治疗的患者还可接受放射治疗和/或可经历手术。
为了预防或治疗疾病,治疗组合物(例如,pgk1、mek/erk、egfr和/或pin1抑制剂)的合适剂量取决于如以上限定的待治疗疾病的类型、疾病的严重程度和进程、施用药剂是为了预防还是治疗目的、先前的治疗、患者临床病史和对药剂的反应和医师的判断。以一次或一系列的治疗向患者适当地施用药剂。
iv实施方案的治疗剂
实施方案的某些方面涉及向确定包含一种或更多种实施方案的生物标志物的患者施用靶向治疗。在一些方面中,向被鉴定为具有表达活化的pgk2(或其生物标志物)的癌症的患者施用pgk1抑制剂、mek/erk抑制剂、egfr抑制剂或pin1抑制剂治疗中的一种或更多种。以下提供了根据实施方案使用的一些特异性靶向治疗。
a.mek/erk激酶抑制剂
可在实施方案的某些方面中使用mek抑制剂,其包括促分裂原活化的蛋白激酶激酶(mapk/erk激酶或mek)或其相关信号途径(如mapk级联反应)的抑制剂。促分裂原活化的蛋白激酶激酶(sic)是使促分裂原活化的蛋白激酶磷酸化的激酶酶。其还被称为map2k。细胞外刺激通过信号级联(“mapk级联”)导致map激酶活化,所述信号级联由map激酶、map激酶激酶(mek、mkk、mekk或map2k)和map激酶激酶激酶(mkkk或map3k)构成。
本文中的mek抑制剂通常是指mek抑制剂。因此,mek抑制剂是指蛋白激酶的mek家族成员的任何抑制剂,包括mek1、mek2和mek5。还提及了mek1、mek2和mek5抑制剂。本领域已知的合适的mek抑制剂的实例包括mek1抑制剂pd184352和pd98059、mek1和mek2抑制剂u0126和sl327以及davies等(2000)中讨论的那些。
特别地,当与其他已知的mek抑制剂相比时,已发现pd184352和pd0325901具有高度的特异性和效力(bain等,2007)。zhang等(2000)中描述了其他的mek抑制剂和mek抑制剂类别。
mek的抑制剂可包括抗体、显性负变体以及抑制mek表达的sirna和反义核酸。mek抑制剂的具体实例包括但不限于:pd0325901(参见例如,rinehart等,2004)、pd98059(例如,可从cellsignalingtechnology得到)、u0126(例如,可从cellsignalingtechnology得到)、sl327(例如,可从sigma-aldrich得到)、arry-162(例如,可从arraybiopharma得到)、pd184161(参见例如,klein等,2006)、pd184352(ci-1040)(参见例如,mattingly等,2006)、舒尼替尼(azd6244/arry-142886/arry-886;参见例如,voss等,us2008/004287通过引用并入本文)、索拉非尼(参见上文voss)、凡德他尼(参见上文voss)、帕唑帕尼(参见例如上文voss)、阿西替尼(参见上文voss)、ptk787(参见上文voss)、refametinib(bay-86-9766/rdea-119)、pimasertib(也称为as703026或msc1936369b)和曲美替尼(trametinib)(gsk-1120212)。
目前,若干种mek抑制剂正在经历临床试验评价。ci-1040已经在癌症的i期和ii期临床试验中进行了评价(参见例如,rinehart等,2004)。正在评价(例如,在临床试验中)的其他mek/erk抑制剂包括pd184352(参见例如english和cobb,2002)、bay43-9006(参见例如chow等,2001)、pd-325901(也称为pd0325901)、arry-438162、rdeal19、rdea-436、ro5126766、xl518、azd8330(也称为arry-704)、gdc-0973、rdea119、pd18416、sch900353、rg-7167、wx-554、e-6201、as-703988、bi-847325、tak-733、rg-7304或fr180204。
b.pin1抑制剂
实施方案的另外一些方面涉及pin1抑制剂以及此类抑制剂的施用。pin1的抑制剂的实例包括但不限于tme-001(2-(3-氯-4-氟-苯基)-异噻唑-3-酮;参见mori等,2011)、5’-硝基-靛玉红肟(indirubinoxime)(yoon等,2012)和环己基酮底物类似物抑制剂,例如ac-pser-ψ[c=och]-pip-色胺(xu等,2012)。xu等(2011)还描述了具有r-pser-ψ[ch2n]-pro-2-(吲哚-3-基)乙胺结构的pin1抑制剂,其中r为芴基甲氧基羰基(fmoc)或ac。肽(例如二硫化物-环化肽)也已被证明是有效的pin1抑制剂,并且可根据本发明实施方案进行使用(参见例如,duncan等(2011),通过引用并入本文)。
c.另外的靶向抑制剂
使用靶向抑制性rna治疗(例如,通过向特定基因或途径施用或表达微小rna(mirna)或小干扰rna(sirna))同样可实现靶向抑制。使用rna介导的干扰可方便地实现例如pgk1、mek/erk、egfr或pin1的抑制。通常来说,将与靶mrna的全部或一部分互补的双链rna分子引入癌细胞,从而促进mrna分子的特异性降解。该转录后机制导致靶向mrna和相应编码蛋白质的表达降低或消除。
此外,已知了用于鉴定新的靶向抑制剂(包括,例如pgk1、mek/erk、egfr或pin1抑制剂)的许多测定。例如,davies等(2000)描述了其中在肽底物和放射性标记的atp存在下孵育激酶的激酶测定。通过激酶磷酸化底物导致将标记并入底物。将每个反应的等分试样固定化在磷酸纤维素纸上,并在磷酸中洗涤以除去游离atp。然后测量孵育后的底物活性并提供激酶活性的指示。使用这种测定可容易地确定候选激酶抑制剂存在和不存在下的相对激酶活性。downey等(1996)还描述了可用于鉴定根据一些实施方案可使用的激酶抑制剂的激酶活性的测定。
d.前药
化合物(例如本实施方案的靶向抑制剂)还可以以前药的形式存在。由于已知前药提高药物的许多期望的品质(例如,溶解性、生物利用度、制备等),因此如果期望的话,本发明的一些方法中使用的化合物可以以前药的形式递送。通常来说,这样的前药将是实施方案的代谢途径抑制剂的功能性衍生物。例如在“designofprodrugs”,编辑h.bundgaard,elsevier,1985;huttunen等,2011;以及hsieh等,2009中描述了用于选择和制备合适的前药衍生物的常规方法,其各自通过引用整体并入本文。
前药可以是生物活性抑制剂(“母体药物”或“母体分子”)的药理上无活性的衍生物,其需要在体内转化以释放活性药物,并且相对于母体药物分子具有改善的递送特性。体内的转化可以是例如一些代谢过程的结果,例如羧酸酯、磷酸酯或硫酸酯的化学或酶水解,或者敏感功能性的还原或氧化。因此,实施方案中使用的化合物的前药可通过修饰化合物中存在的官能团进行制备,以这样的方式使得修饰物在常规操作中或在体内切割为母体化合物。前药包括例如其中羟基、氨基或羧基键合到任何基团的本文中所述的化合物,使得将当前药向对象施用时,所述化合物切割以分别形成羟基、氨基或羧酸。因此,本发明考虑本发明化合物的前药以及递送前药的方法。
e.抑制性寡核苷酸
抑制性寡核苷酸可抑制细胞中基因的转录或翻译。寡核苷酸可以是5至50个或更多个核苷酸长,并且在某些实施方案中为7至30个核苷酸长。在某些实施方案中,寡核苷酸可以是7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸长。寡核苷酸可包含核酸和/或核酸类似物。通常来说,抑制性寡核苷酸将抑制细胞内单个基因(例如,pgk1)的翻译;然而,在某些实施方案中,抑制性寡核苷酸可抑制细胞内的多于一种基因的翻译。
在寡核苷酸内,寡核苷酸的组分不需要全部是相同类型或同质的(例如,寡核苷酸可包含核苷酸和核酸或核苷酸类似物)。在本发明的某些实施方案中,寡核苷酸可仅包含单个核酸或核酸类似物。抑制性寡核苷酸可包含与互补核酸杂交以形成双链结构的5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30个或更多个连续的核酸碱基(nucleobase),包括其间的所有范围。
iii组合治疗
包括组合治疗的方法和组合物提高治疗和保护效果,和/或提高其他抗癌或抗增殖治疗的治疗效果。治疗和预防方法和组合物可以以有效实现期望效果(例如杀伤癌细胞和/或抑制细胞过度增殖)的组合量来提供。组织、肿瘤或细胞可与包含一种或更多种药剂的一种或更多种组合物或药物制剂接触,或者通过使组织、肿瘤和/或细胞与两种或更多种不同的组合物或制剂接触。此外,考虑这样的组合治疗可与放射治疗、手术治疗或免疫治疗结合使用。
组合施用可包括同时施用两种或更多种相同剂型的药剂、同时施用不同的剂型以及分开施用。即,该主题治疗组合物与其他治疗剂可以以相同剂型一起配制并同时施用。或者,主题治疗组合物与其他治疗剂可同时施用,其中两种药剂均存在于单独的制剂中。在另一个替代方案中,可在其他治疗剂之后紧接着施用治疗剂,反之亦然。在分开的施用方案中,主题治疗组合物和其他治疗剂可以间隔几分钟、或间隔几小时、或间隔几天施用。
第一抗癌治疗可在相对于第二抗癌治疗之前、期间、之后或以多种组合施用。施用可以以同时至间隔数分钟至数天至数周进行。在其中向患者分开提供第一治疗与第二治疗的实施方案中,通常将确保在每次递送时间之间的相当长的一段时间内不会失效,使得两种化合物仍然能够对患者发挥有利的组合效应。在这种情况下,考虑在彼此相隔约12小时至24小时或72小时内,更特别地在相隔约6小时至12小时内可为患者提供第一治疗和第二治疗。在一些情况下,可能期望显著地延长治疗的时间周期,其中各自施用之间的间隔由数天(2、3、4、5、6或7)延长至数周(1、2、3、4、5、6、7或8)。
在某些实施方案中,治疗过程将持续1至90天或更长(该范围包括间隔天数)。考虑可在第1天至第90天(该范围包括间隔天数)或其任意组合的任一天给予一种药剂,并且在第1天至第90天(该范围包括间隔天数)或其任意组合的任一天给予另一种药剂。在一天内(24小时期间),可给予患者一次或多次药剂施用。此外,在治疗过程之后,考虑存在不施用抗癌治疗的一段时间。该时间段可持续1至7天,和/或1至5周,和/或1至12个月或更长(该范围包括间隔天数),这取决于患者的状况,例如其预后、强度、健康等。预期如果必要将重复治疗周期。
可以使用多种组合。例如,以下pgk1、mek/erk、egfr、和/或pin1抑制剂为“a”,且另一种抗癌治疗为“b”。
a/b/ab/a/bb/b/aa/a/ba/b/bb/a/aa/b/b/bb/a/b/b
b/b/b/ab/b/a/ba/a/b/ba/b/a/ba/b/b/ab/b/a/a
b/a/b/ab/a/a/ba/a/a/bb/a/a/aa/b/a/aa/a/b/a
考虑到药剂的毒性(如果有的话),向患者施用本发明的任何化合物或治疗将遵循用于施用此类化合物的一般方案。因此,在一些实施方案中,存在可归因于组合治疗的监测毒性的步骤。
a.化学治疗
根据本发明可使用多种化学治疗剂。术语“化学治疗”是指使用药物来治疗癌症。“化学治疗剂”用于意指在癌症治疗中施用的化合物或组合物。通过这些药剂或药物在细胞内的活性模式(例如,它们是否影响细胞周期或在哪个阶段影响细胞周期)对其进行分类。或者,可以基于药剂直接交联dna、插入dna或通过影响核酸合成来诱导染色体和有丝分裂畸变的能力来对其进行表征。
化学治疗剂的实例包括烷化剂,例如噻替派和环磷酰胺;烷基磺酸酯,例如白消安、英丙舒凡和哌泊舒凡;氮丙啶类,例如苯佐替哌(benzodopa)、卡波醌、美妥替哌(meturedopa)和乌瑞替哌(uredopa);乙撑亚胺类(ethylenimines)和甲基蜜胺类(methylamelamines),包括六甲蜜胺、三乙撑蜜胺(triethylenemelamine)、三乙撑磷酰胺(trietylenephosphoramide)、三乙撑硫代磷酰胺和三羟甲基蜜胺;番荔枝内酯类(acetogenin)(尤其是布拉它辛(bullatacin)和布拉它辛酮(bullatacinone));喜树碱(包括合成类似物拓扑替康);苔藓抑素;callystatin;cc-1065(包括其阿多来新、卡折来新和比折来新合成类似物);隐藻素类(cryptophycin)(特别是隐藻素1和隐藻素8);多拉司他丁;倍癌霉素(duocarmycin)(包括合成类似物kw-2189和cb1-tm1);艾榴塞洛素(eleutherobin);水鬼蕉碱(pancratistatin);沙考地汀(sarcodictyin);海绵抑素(spongistatin);氮芥类,例如苯丁酸氮芥、萘氮芥、氯膦酰胺(cholophosphamide)、雌莫司汀(estramustine)、异环磷酰胺、双氯乙基甲胺(mechlorethamine)、盐酸氧氮芥、美法仑、新氮芥(novembichin)、苯芥胆甾醇、泼尼莫司汀(prednimustine)、曲磷胺和尿嘧啶氮芥;亚硝基脲类(nitrosureas),例如卡莫司汀、氯脲菌素、福莫司汀、洛莫司汀、尼莫司汀和雷莫司汀(ranimnustine);抗生素类,例如烯二炔类抗生素(enediyneantibiotic)(例如,加利车霉素(calicheamicin),尤其是加利车霉素γ1i和加利车霉素ωi1;达内霉素(dynemicin),包括达内霉素a;二膦酸盐类(bisphosphonate),例如氯屈膦酸盐(clodronate);埃斯波霉素(esperamicin);以及新抑癌菌素(neocarzinostatin)生色团及相关的色蛋白烯二炔类抗生素生色团、阿克拉霉素(aclacinomysin)、放线菌素、氨茴霉素(authrarnycin)、重氮丝氨酸(azaserine)、博来霉素、放线菌素c(cactinomycin)、卡洛比星(carabicin)、洋红霉素、嗜癌霉素(carzinophilin)、色霉素(chromomycinis)、放线菌素d(dactinomycin)、柔红霉素、地托比星、6-重氮-5-氧代-l-正亮氨酸、多柔比星(包括吗啉代-多柔比星、氰基吗啉代-多柔比星、2-吡咯啉基-多柔比星和脱氧多柔比星)、表柔比星、伊索比星、伊达比星(idarubicin)、麻西罗霉素(marcellomycin)、丝裂霉素(例如丝裂霉素c)、霉酚酸、诺加霉素(nogalarnycin)、橄榄霉素、培洛霉素(peplomycin)、泊非霉素(potfimromycin)、嘌呤霉素、三铁阿霉素、罗多比星、链黑菌素、链佐星(streptozocin)、杀结核菌素、乌苯美司、净司他丁和佐柔比星;抗代谢物类,例如甲氨蝶呤和5-氟尿嘧啶(5-fu);叶酸类似物,例如二甲叶酸、蝶罗呤和三甲曲沙(trimetrexate);嘌呤类似物,例如氟达拉滨、6-巯基嘌呤、硫咪嘌呤(thiamiprine)和硫鸟嘌呤;嘧啶类似物,例如安西他滨、阿扎胞苷、6-氮尿苷、卡莫氟、阿糖胞苷、二脱氧尿苷、脱氧氟尿苷、依诺他滨和氟尿苷;雄激素,例如卡鲁睾酮、丙酸甲雄烷酮(dromostanolonepropionate)、环硫雄醇(epitiostanol)、美雄烷和睾内酯;抗肾上腺,例如米托坦(mitotane)和曲洛司坦;叶酸补充剂,例如亚叶酸(frolinicacid);醋葡醛内酯;醛磷酰胺糖苷;氨基乙酰丙酸;恩尿嘧啶;安吖啶;贝塔布辛(bestrabucil);比生群(bisantrene);依达曲沙(edatraxate);地磷酰胺(defofamine);地美可辛;地吖醌(diaziquone);伊弗尼辛(elformithine);醋酸羟吡咔唑(elliptiniumacetate);埃博霉素(epothilone);依托格鲁(etoglucid);硝酸镓;羟基脲;香菇多糖(lentinan);氯尼达明(lonidainine);美登素生物碱类(maytansinoid),例如美登素和安丝菌素;米托胍腙;米托蒽醌;莫哌达醇(mopidanmol);尼曲吖啶(nitraerine);喷司他丁;蛋氨氮芥(phenamet);吡柔比星;洛索蒽醌;鬼臼酸(podophyllinicacid);2-乙基酰肼;丙卡巴肼;psk多糖复合物);雷佐生(razoxane);根毒素(rhizoxin);西索菲兰(sizofiran);锗螺胺(spirogermanium);细交链孢菌酮酸;三亚胺醌(triaziquone);2,2′,2″-三氯三乙胺;单端孢霉烯类(trichothecene)(尤其是t-2毒素、疣孢菌素a(verracurina)、杆孢菌素a和蛇形菌素(anguidine));乌拉坦(urethan);长春地辛;达卡巴嗪;甘露氮芥(mannomustine);二溴甘露醇;二溴卫矛醇;哌泊溴烷(pipobroman);甲托辛(gacytosine);阿糖胞苷(“ara-c”);环磷酰胺;紫杉烷类,例如紫杉醇和多西他赛(doxetaxel);吉西他滨;6-硫鸟嘌呤;巯基嘌呤;铂配位复合物,例如顺铂、奥沙利铂和卡铂;长春碱;铂;依托泊苷(vp-16);异环磷酰胺;米托蒽醌;长春新碱;长春瑞滨;诺肖灵(novantrone);替尼泊苷;依达曲沙(edatrexate);柔红霉素;氨基蝶呤;希罗达(xeloda);伊班膦酸(ibandronate);伊立替康(例如,cpt-11);拓扑异构酶抑制剂rfs2000;二氟甲基鸟氨酸(dmfo);类视黄醇a,例如视黄酸;卡培他滨;卡铂、丙卡巴肼、普卡霉素(plicomycin)、吉西他滨(gemcitabien)、诺维本(navelbine)、法呢基蛋白转移酶抑制剂、反铂(transplatinum);以及上述任一种的可药用盐、酸或衍生物。
b.放射治疗
导致dna损伤并已广泛使用的其他因素包括通常被称为γ射线、x射线的那些和/或向肿瘤细胞定向递送放射性同位素。还考虑了其他形式的dna损伤因素,例如微波、质子束照射(美国专利5,760,395和4,870,287)和uv辐照。最有可能所有这些因素对dna、dna前体、dna的复制和修复以及染色体的组装和维持引起宽范围的损害。x射线的剂量范围以50至200伦琴日剂量持续一段长时间(3至4周)至2000至6000伦琴的单剂量。放射性同位素的剂量范围变化很大,且取决于同位素的半衰期、放射辐射的强度和类型以及赘生性细胞的摄取。
c.免疫治疗
本领域技术人员将理解,另外的免疫治疗可与本发明的方法结合或组合使用。在癌症治疗的背景下,免疫治疗通常依赖于使用免疫效应细胞和分子来靶向并破坏癌细胞。利妥昔单抗(
在免疫治疗的一个方面中,肿瘤细胞必须具有进行靶向的一些标志物,即,不存在于大多数其他细胞上。存在许多肿瘤标志物,并且这些肿瘤标志物中的任何一种可能适于本发明的上下文中的靶向。常见的肿瘤标志物包括cd20、癌胚抗原、酪氨酸酶(p97)、gp68、tag-72、hmfg、唾液酸化的路易斯抗原、muca、mucb、plap、层黏连蛋白受体、erbb和p155。免疫治疗的一个替代方面是将抗癌作用与免疫刺激作用相结合。还存在免疫刺激分子,包括:细胞因子,例如il-2、il-4、il-12、gm-csf、γ-ifn,趋化因子例如mip-1、mcp-1、il-8和生长因子,例如flt3配体。
目前正在研究或正在使用的免疫治疗的实例是免疫佐剂,例如,牛分枝杆菌(mycobacteriumbovis)、恶性疟原虫(plasmodiumfalciparum)、二硝基氯苯和芳族化合物(美国专利5,801,005和5,739,169;hui和hashimoto,1998;christodoulides等,1998);细胞因子治疗,例如干扰素α、β和γ、il-1、gm-csf和tnf(bukowski等,1998;davidson等,1998;hellstrand等,1998);基因治疗,例如tnf、il-1、il-2和p53(qin等,1998;austin-ward和villaseca,1998;美国专利5,830,880和5,846,945);以及单克隆抗体,例如抗cd20、抗神经节苷脂gm2和抗p185(hanibuchi等,1998;美国专利5,824,311)。考虑一种或更多种抗癌治疗可与本文中所述的抗体治疗一起使用。
d.手术
约60%患有癌症的人将经历某种类型的手术,其包括预防性、诊断性或分期、治疗性和缓解性手术。治愈性手术包括其中全部或一部分癌组织被物理去除、切除和/或破坏的切除术,并且可以与其他治疗(例如本发明的治疗、化学治疗、放射治疗、激素治疗、基因治疗、免疫治疗和/或替代治疗)结合使用。肿瘤切除术是指物理去除至少一部分肿瘤。除肿瘤切除术之外,通过手术的治疗包括激光手术、冷冻手术、电外科手术和用显微镜控制的手术(莫氏手术(mohs’surgery))。
在切除一部分或全部癌性细胞、组织或肿瘤之后,可以在体内形成腔。治疗可通过灌注、直接注射或向该区域局部施用另外的抗癌治疗来实现。这样的治疗可以重复,例如每1、2、3、4、5、6或7天,或者每1、2、3、4和5周,或者每1、2、3、4、5、6、7、8、9、10、11或12个月。这些治疗也可具有不同的剂量。
e.另外的药剂
考虑可以将另外的药剂与本发明的某些方面组合使用以改善治疗的治疗效力。这些另外的药剂包括影响细胞表面受体与gap连接的增量调节的药剂、细胞抑制剂和分化剂、细胞黏附抑制剂、提高过度增殖细胞对细胞凋亡诱导剂或其他生物药剂的敏感性的试剂。通过提高gap连接数提高细胞间信号转导将会提高相邻过度增殖细胞群的抗过度增殖性效应。在另一些实施方案中,细胞抑制剂或分化剂可与本发明的某些方面组合使用以改善治疗的抗过度增殖效力。考虑细胞黏附抑制剂以改善本发明的效力。细胞黏附抑制剂的实例是黏着斑激酶(focaladhesionkinase,fak)抑制剂和洛伐他汀。进一步考虑提高过度增殖细胞对细胞凋亡的敏感性的其他试剂(例如抗体c225)可以与本发明的某些方面组合使用以提高治疗效力。
iv.药盒和诊断
在本发明的多个方面中,预期包含诊断剂、治疗剂和/或其他治疗剂和递送剂的药盒(kit)。在一些实施方案中,本发明考虑用于制备和/或施用本发明治疗的药盒。药盒可包含能够用于施用本发明的活性剂或有效剂的试剂。药盒的试剂可包含至少一种基因表达的抑制剂,组合治疗的一种或更多种抗癌组分,以及用于制备、配制和/或施用本发明的组分或者实施本发明方法的一个或更多个步骤的试剂。
在一些实施方案中,药盒还可包含合适的容器设备,其是不与药盒的组分起反应的容器,例如eppendorf管、测定板、注射器、瓶或管。容器可以由可灭菌材料(例如塑料或玻璃)制成。
药盒还可包含概述方法之程序步骤的说明书,其将遵循与本文中所述基本上相同的程序或本领域普通技术人员已知的程序。
v.实施例
包括以下实施例以说明本发明的一些优选实施方案。本领域技术人员应理解,以下实施例中公开的技术表示发明人发现的在本发明的实践中运行良好的技术,并因此可认为构成用于其实施的优选模式。然而,根据本公开内容,本领域技术人员应理解,在不脱离本发明的精神和范围的情况下,可以对所公开的具体实施方案中进行许多改变,并且仍然获得相似或类似的结果。
材料和方法
材料。识别pgk1、pgk138-qrikaa-43、磷酸-pgk1s203、pdhk1、磷酸-pdhk1t338、磷酸-pdhk1y243、egfr、磷酸-egfry1172和ha的兔多克隆抗体获自signalwayantibody(collegepark,maryland)。鱼藤酮,识别pgk1、pdhk1、coxiv、timm22、v5、pdhe1α、pdhe1αps293的兔多克隆抗体以及识别具有天然构象的兔igg的小鼠单克隆抗体获自abcam(cambridge,ma)。识别mapk/apk2、mapk/apk2pt222、c-jun和c-junps73的兔多克隆抗体购自cellsignalingtechnology(danvers,ma)。识别hif1α、细胞色素c和磷酸丝氨酸的小鼠抗体获自bdbiosciences(bedford,ma)。针对gst、微管蛋白、erk1/2、perk1/2、pin1和磷酸-苏氨酸的单克隆抗体购自santacruzbiotechnology(santacruz,ca)。活性甘油醛3-磷酸脱氢酶(gapdh)重组蛋白、flag的小鼠单克隆抗体、tritonx-100、α-胰凝乳蛋白酶、甘油醛3-磷酸、氯化锂、丙酮酸、苹果酸、egta、atpase、egf、二氯乙酸盐(dca)、磷酸烯醇丙酮酸、丙酮酸激酶、乳酸脱氢酶、adp、nadh和nad+购自sigma(st.louis,mo)。识别mnsod和tom20的兔多克隆抗体、u0126、sp600125、sb203580、ag1478、嘌呤霉素、杀稻瘟菌素和无dnase的rnasea购自emdbiosciences(sandiego,ca)。蛋白酶k和mitotrackerredcmxros购自invitrogen(carlsbad,california)。针对tom20和ki67的单克隆抗体购自millipore(sandiego,ca)。hyfect转染试剂来自denvillescientific(metuchen,nj)。纯化的his-pdhe1α重组蛋白质来自signalchem(richmond,canada)。免疫金标记的抗兔二抗购自tedpella(redding,ca)。含有s203/p204基序的pgk1的合成磷酸化的(hhhhhhlepsper-pna;seqidno:1)和非磷酸化的(hhhhhhlesper-pna[seqidno:1]和hhhhhhledper-pna[seqidno:2])寡肽购自selleckchemicals(houston,tx)。[γ32p]atp购自mpbiochemicals(santaana,ca)。[1-14c]-丙酮酸购自americanradiolabeledchemicals(st.louis,mo)。dapi、alexafluor488山羊抗兔抗体、海胺氢氧化物(hyaminehydroxide)、闪烁管和靶向hif1α的sirna购自thermofisherscientific(pittsburgh,pa)。链霉亲和素珠购自pierce(rockford,il)。
细胞和细胞培养条件。将u87、u87/egfr和u251人gbm细胞、bxpc-3人胰腺癌细胞、chl1人黑素瘤细胞和小鼠胚胎成纤维细胞(mouseembryofibroblast,mef)维持在不含丙酮酸的补充有10%经透析的小牛血清(hyclone,logan,ut)的dulbecco改良的eagle培养基(dmem)中。将人原代gsc11gbm细胞维持在补充有b27(invitrogen,carlsbad,california)、表皮生长因子(10ng/ml)和碱性成纤维细胞生长因子(10ng/ml)的dmem/f-1250/50中。细胞在正常氧(20%氧气)或缺氧(1%氧气)的条件下培养。对于egf处理,通过使细胞培养物生长至汇合而使其静止,并每天用含有0.5%血清的新鲜培养基更换培养基。使用终浓度为100ng/ml的egf进行细胞刺激。
转染。在转染前18小时,以4×105/60mm平皿的密度将细胞进行平板接种。如前所述进行转染(xia等,2007)。
亚细胞分级。使用来自biovision(mountainview,ca)的线粒体/胞质溶胶分级试剂盒来分离线粒体级分和胞质溶胶级分。线粒体蛋白质和胞质溶胶蛋白质用于免疫印迹分析。对于蛋白酶k保护测定,使线粒体沉淀重悬于低张缓冲液(10mmhepes-koh[ph7.4]和1mmedta)中,并用含有或不含1%tritonx-100或毛地黄皂苷(100μm)的蛋白酶k(200mg/ml)在冰上消化20分钟。然后将消化物用三氯乙酸沉淀,并用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sds-page)和免疫印迹进行分析。如先前所述进行线粒体亚分级(subfractionation)(she等,2011)。简言之,将经分离的线粒体重悬于10μmkh2po4(ph7.4)中在冰上保持20分钟。添加等体积的等渗溶液(32%蔗糖、30%甘油、10mmmgcl2),并在10,000g和4℃下离心10分钟。将上清液在15,000g和4℃下离心1小时;沉淀和上清液分别含有外膜蛋白质和膜间隙蛋白质。然后,将来自第一次离心的沉淀重悬于10μmkh2po4(ph7.4)中在冰上保持20分钟,添加等渗溶液,然后在15,000g和4℃下离心1小时;沉淀和上清液分别含有内膜蛋白质和基质蛋白。
线粒体[1-14c]-丙酮酸转化测定。按照先前所述(pezzato等,2009)并稍加修改进行[1-14c]-丙酮酸转化测定。简言之,使用分离的线粒体(1mg)在1ml含有[1-14c]-丙酮酸(0.1μci/ml)的线粒体重悬缓冲液(200mm蔗糖、10mmhepes[ph7.4]、5mm苹果酸、2mm磷酸二氢钠、2mmadp、1mmegta)中测量通过pdh复合物的14co2产生。将2mleppendorf管中的孵育混合物置于带有箔衬里螺帽的20ml闪烁管的底部并维持搅拌。孵育期间产生的14co2在闪烁管的底部被1ml海胺氢氧化物捕获。将反应混合物从闪烁管中移出并用0.5ml的50%三氯乙酸封闭1小时。然后,向每个闪烁管中添加19ml闪烁液(scintillatorliquid),并在闪烁计数器上测量放射性。使用牛血清白蛋白作为标准,基于通过bradford测定法测量的线粒体蛋白质水平将结果归一化。
免疫金透射电子显微术。将固定的样品在0.1m二甲砷酸盐缓冲液中洗涤,并用0.1%微孔过滤缓冲的单宁酸进行处理,随后用1%缓冲的四氧化锇固定30分钟,并用1%微孔过滤的乙酸双氧铀(uranylacetate)整体(enbloc)染色。将样品在水中洗涤若干次,在浓度递增的乙醇中进行脱水,浸润并包埋在lx-112培养基(laddresearch,williston,vt)中。使样品在60℃烘箱中聚合2天。用leicaultracut切片机(deerfield,il)切割超薄切片,在leicaem染色机中用乙酸双氧铀和柠檬酸铅染色,并用jem1010透射电子显微镜(jeol,usa,inc.,peabody,ma)在80kv的加速电压下检查。使用先进显微技术成像系统(advancedmicroscopytechniquesimagingsystem)(danvers,ma)获得数字图像。
体外异构化测定。用通过异构体特异性蛋白质水解测定的顺式肽含量显示异构化速率。通过将肽与α-胰凝乳蛋白酶在0℃下孵育2分钟以完全水解4-硝基苯胺键处的反式异构体以获得纯的顺式肽来制备顺式肽。使纯的顺式肽保持重新平衡。随着异构化的进行,在指定的时间取出等分试样。添加胰凝乳蛋白酶抑制剂(chymostatin)以灭活胰凝乳蛋白酶。在390nm处测量释放的4-硝基苯胺的吸光度。
ros的测量。将收获的细胞在td缓冲液(25mmtris[ph7.4]、5mmkcl、400μmna2hpo4和150mmnacl)中洗涤,然后在37℃下,在含有5mmmitosox(红色线粒体超氧化物指示剂)(invitrogen,m36008)的td缓冲液中重悬15分钟。用td缓冲液进一步洗涤后,将细胞在96孔板上进行平板接种,并且用synergyht微孔板读数器(biotek,winooski,vt)在485/590nm处进行测量。将重悬的分离的线粒体在含有不同浓度的丙酮酸的缓冲液(200mm蔗糖、10mmhepes[ph7.4]、5mm苹果酸、2mm磷酸二氢钠、2mmadp、1mmegta)中在20℃下孵育30分钟。根据对所收获细胞的描述进行线粒体ros的测量。
线粒体膜电位的测量。将分离的线粒体(1mg)在含有不同浓度的丙酮酸的重悬缓冲液(200mm蔗糖、10mmhepes[ph7.4]、5mm苹果酸、2mm磷酸二氢钠、2mmadp、1mmegta)中在20℃下孵育30分钟。使用线粒体膜电位测定试剂盒(abcam,cambridge,ma)测量线粒体膜电位。
乳酸产生的测量。将细胞接种在培养皿中,6小时后用无血清dmem更换培养基。将细胞孵育6小时,然后收集培养基用于测量乳酸浓度。使用biovision乳酸测定试剂盒(biovision,milpitas,ca)来确定乳酸水平。
胞质溶胶丙酮酸和线粒体乙酰辅酶a浓度的测量。将细胞接种在培养皿中并在正常氧或缺氧期间培养6小时。通过使用丙酮酸比色测定试剂盒(biovision)和乙酰辅酶a荧光测定试剂盒(biovision)分别测量胞质溶胶丙酮酸和线粒体乙酰辅酶a的浓度。
pgk1活性的测量。在96孔板中,将经纯化的重组wt或突变体pgk1(1ng)在100μl反应缓冲液(5mmkh2po4、ph7.0、1mmgap、0.3mmβ-nad、0.2mmadp、5mmmgso4、100mm甘氨酸、5ng/μlgapdh)中在25℃下孵育,并且在动力学模式下在339nm读取5分钟。
质谱分析。用200ng测序级改良的胰蛋白酶(promega,madison,wi)将经纯化的pdhk1的体外pgk1磷酸化的样品在含有rapigest(waterscorp.,milford,ma)的50mm碳酸氢铵缓冲液中在凝胶中于37℃下消化过夜。在obitrap-elite质谱仪(thermofisherscientific,waltham,ma)上通过lc-ms/ms分析消化物。通过proteomediscoverersoftwarev.1.4(thermofisherscientific),使用mascotserverv.2.3(matrixscience,london,uk)和sequestv.1.27(universityofwashington,seattle,wa),通过针对swissprot蛋白质数据库(ebi)的片段谱的数据库检索来鉴定蛋白质。通过使用在proteomediscoverer中执行的phosphors算法来分析磷酸肽匹配并进行人工验证(curated)(taus等,2011)。
免疫沉淀和免疫印迹分析。用改良的缓冲液从培养的细胞中提取蛋白质,随后如先前所述用相应的抗体进行免疫沉淀和免疫印迹(lu等,1998)。
链霉亲和素和gst拉下测定。将链霉亲和素或谷胱甘肽琼脂糖珠与细胞裂解物(1mg/ml)或经纯化的蛋白质孵育12小时。将所述珠用裂解物缓冲液洗涤三次。
细胞增殖测定。在缺氧期间将共计2×105个细胞平板接种并在接种至具有10%经透析的小牛血清的dmem中之后4天进行计数。数据表示3个独立实验的平均值±sd。
dna构建体和诱变。将聚合酶链反应(pcr)扩增的人pgk1、tom20、pdhk1、pdhk2、pdhk3、pdhk4、k-rasg12v或b-rafv600e克隆至pcoldi、pgex-4t-1、pe-sumo、pcdna6/hisv5或pcdna6/sfb载体。pcdna6/hisv5pdhk1t338a、pe-sumopdhk1t338a、pgex-4t-1pgk1s203a、pgex-4t-1pgk1s203d、pgex-4t-1pgk1r39/k41a、pgex-4t-1pgk1s203dr39/k41a、pcoldipgk1s203a、pcoldipgk1s203d、pcoldipgk1t378p、pcoldipgk1r39/k41a、pcdna6/hisv5pgk1s203a、pcdna6/hisv5pgk1s203d、pcdna6/hisv5pgk1t378p、pcdna6/hisv5pgk1r39/k41a、pcdna6/hisv5pgk1k48a、pcdna6/sfbpgk1r39/k41a、pcdna6/sfbpgk1k48a、pcdna6/sfbpgk1v81/83a、pcdna6/sfbpgk1v177/179a和pcdna6/sfbpgk1v278a/i280r/l282r通过使用quickchange定点诱变试剂盒(stratagene,lajolla,ca)来制备。pcdna6/hisv5rpgk1含有c652g、t655c、c658g和t661c的无义突变。pcdna6/hisv5rpdhk1含有c848t、a850g、a853g、c854t和t856g的无义突变。
用对照寡核苷酸gcttctaacaccggaggtctt(seqidno:3)形成pgipz对照。分别用ggatgtctatgtcaatgatgc(seqidno:4)和ccgaactagaacttgaaga(seqidno:5)形成pgipzpgk1shrna和pdhk1shrna。
重组蛋白质的纯化。如先前所述使wt和突变体his-pgk1、gst-pgk1、his-tom20、his-pin1、gst-pin1和sumo-pdhk1在细菌中表达并纯化(xia等,2007)。
体外激酶测定和磷酸氨基酸分析(paa)。如先前所述进行激酶反应(fang等,2007)。简言之,将经细菌纯化的重组pgk1(10ng)与pdhk1(100ng)在25μl激酶缓冲液(50mmtris-hcl[ph7.5]、100mmkcl、50mmmgcl2、1mmna3vo4、1mmdtt、5%甘油、0.5mmatp和10μci[γ32p]atp)中在25℃下孵育1小时。对于使用1,3-二磷酸甘油酸酯(1,3-bpg)作为磷酸供体的测定,将活性甘油醛-3-磷酸脱氢酶(200ng)与pgk1(100ng)和pdhk1(100ng)在25μl反应缓冲液(5mmkh2po4[ph7.0]、1mmgap、0.3mmβ-nad、5mmmgso4、100mm甘氨酸、1mmna3vo4、1mmdtt)中在25℃下孵育1小时。通过添加sds-page加样缓冲液终止反应并加热至100℃。然后使反应混合物进行sds-page分析。
对于paa,通过sds-page来分离激酶反应混合物,转移到聚偏二氟乙烯膜上,并切除与磷酸化的pdhk1的移动性一致的膜。如先前所述进行在薄层纤维素板上使用二维电泳的paa(vandergeer和hunter,1994)。
免疫荧光分析。如先前所述进行免疫荧光分析(fang等,2007)。
分子建模分析。使用在线zdock服务器(在万维网网址zdock.umassmed.edu/上)进行pgk1和pin1蛋白质的分子对接分析。选择pgk1的s203和pin1(k63和r69)的ww结构域用于对接。
丙酮酸氧化速率的测量。将在具有补充有10%bcs、20mmhepes、5mm葡萄糖和1mm丙酮酸的不含碳酸氢钠的dmem的25-cm2斜颈培养瓶中培养的u87细胞(2×106)不进行缺氧处理或缺氧处理6小时,然后添加0.2μci的[1-14c]-丙酮酸(americanradiolabeledchemicals)。用浸泡在海胺氢氧化物中的滤纸覆盖烧瓶的出口,并在37℃下孵育2小时。移除滤纸,并使用ls6500多功能闪烁计数器(beckmancounter)测定放射性。丙酮酸氧化速率表示为归一化为细胞数目的样品的放射性水平。对于egf处理,将在补充有0.5%bcs、20mmhepes、5mm葡萄糖、1mm丙酮酸的不含碳酸氢钠的dmem的25-cm2斜颈培养瓶中培养的2×106个u87/egfr细胞不用egf处理或用egf(100ng/ml)处理6小时,然后添加0.2μci的[1-14c]-丙酮酸。
氧消耗速率的测量。将细胞(3×104)平板接种到dmem(0.5%bcs、25mm葡萄糖、2mm谷氨酰胺)(seahorsebioscience,northbillerica,ma)中的xf24板上,并在37℃、5%co2下孵育过夜,用5mm二氯乙酸盐预处理1小时,并随后用egf(100ng/ml)刺激6小时或不用egf刺激。然后用补充有2mm谷氨酰胺、25mm葡萄糖(使用0.5mm氢氧化钠将ph调节至7.4)的675μl未缓冲的测定培养基(seahorsebioscience)替换该培养基。然后将细胞置于37℃的无co2的培养箱中30分钟。使用xf24板读数器记录基础氧消耗速率(basaloxygen-consumptionrate,ocr)。计算线粒体ocr(δocr值为1μm的鱼藤酮处理前后的ocr差异),并用细胞数目归一化。
14c-脂质合成测定。测量来源于14c-葡萄糖或14c-谷氨酰胺的14c-脂质。将接种在6孔板上的半汇合(subconfluent)细胞与egf(100ng/ml)预孵育24小时或不与其预孵育。然后将这些细胞在含有1μci的d-[6-14c]-葡萄糖(americanradiolabeledchemicals)或l-[u-14c]-谷氨酰胺(perkinelmer)的新鲜培养基中孵育2小时,然后进行pbs洗涤。通过添加500μl己烷∶异丙醇(3∶2v/v)提取脂质。将细胞与另外500μl己烷∶异丙醇溶液一起孵育。合并脂质提取物并加热空气干燥。将提取的脂质重悬于50μl氯仿中并进行闪烁计数。用细胞数目将闪烁计数归一化。
确定pgk1的km。将经纯化的重组wtpgk1(0.2ng)在100μl具有指定atp浓度的反应缓冲液(50mmtris-hcl[ph7.5]、600ng/μlsumo-pdhk1、5mmmgso4、1mmna3vo4、1mmdtt、1.8mm磷酸烯醇丙酮酸、7单位丙酮酸激酶、10单位乳酸脱氢酶、0.01mmnadh)中在96孔板中于37℃下孵育,并且通过多重检测微板读数器(bmglabtech,cary,nc)在动力学模式下在339nm处读取5分钟。通过测量作为时间之函数的产物浓度来获得反应速度(v)。根据lineweaver-burke绘图模型,由1/v相对于1/[atp]的图计算km。数据表示3个独立实验的平均值±sd。
线粒体基质中atp浓度的测量。将经分离的线粒体(2mg)在洗涤缓冲液(200mm蔗糖、10mmhepes[ph7.4]、100μm毛地黄皂苷)中洗涤,并随后在100μl测定缓冲液中裂解。使用10kda的离心柱(millipore)将样品脱蛋白,并使用biovisionatp测定试剂盒(biovision)测定atp水平。
颅内注射。将具有内源性pgk1或pdhk1耗竭及其wt或突变体蛋白质的重建表达的gbm细胞经颅内注射(5μldmem中1×106个细胞/小鼠)到4周龄的雌性无胸腺裸小鼠中。如先前所述进行注射(gomez-manzano等,2006)。在每个实验中,每组使用7只小鼠。注射有u87或gsc11细胞的动物分别在注射胶质瘤细胞之后28或21天处死。获得每只小鼠的脑,固定在4%甲醛中,并包埋在石蜡中。通过h&e染色切片的组织学分析来确定肿瘤形成和表型。根据相关制度和国家指导方针和条例对动物进行处理。动物的使用经德克萨斯大学md安德森癌症中心机构审查委员会(institutionalreviewboardattheuniversityoftexasmdandersoncancercenter)批准。
组织学评价和免疫组织化学染色。将小鼠肿瘤组织固定并准备用于染色。用mayer的苏木精以及随后用曙红(h&e)(biogenexlaboratories,sanramon,ca)对标本进行染色。此后,使用universalmount(researchgeneticshuntsville,al)固封载玻片。
用针对磷酸pgk1s203、磷酸pdhk1t338、磷酸pdhs293的抗体或非特异性igg作为阴性对照对来自石蜡包埋的人gbm标本的组织切片进行染色。如先前所定义的,根据阳性细胞的百分比和染色强度对组织切片定量评分(ji等,2009)。指定以下比例分数:如果0%的肿瘤细胞显示阳性染色则为0,如果0%至1%则为1,如果2%至10%则为2,如果11%至30%则为3,如果31%至70%则为4,如果71%至100%则为5。以0至3的等级评定染色强度:0,阴性;1,弱;2,中等;以及3,强。如先前所述,将比例和强度评分合并得到总评分(范围,0至8)(ji等,2009)。将评分与总存活(定义为从诊断日期至死亡或最后一次已知的随访日期的时间)进行比较。所有患者在手术后接受标准辅助性放射治疗,随后用烷化剂(多数情况下为替莫唑胺)进行治疗。人脑肿瘤标本和数据库的使用经德克萨斯大学md安德森癌症中心机构审查委员会(institutionalreviewboardattheuniversityoftexasmdandersoncancercenter)批准。
tunel测定。小鼠肿瘤组织被切成5μm厚度。根据制造商的说明书,通过使用deadendtmtunelsystems(promega,madison,wi)来检测凋亡细胞。
实施例1-通过erk1/2依赖性pgk1s203磷酸化介导缺氧以及egfr、k-ras和b-raf的活化诱导的pgk1线粒体易位
在实体瘤中,缺氧似乎与肿瘤生长和发展密切相关(koppenol等,2011;hockel和vaupel,2001;caims等,2011)。为了测试糖酵解途径中的atp生成酶pgk1是否具有任何亚细胞区室依赖性功能,将细胞暴露于缺氧环境并检测pgk1的细胞分布。将u87人胶质母细胞瘤(gbm)细胞在缺氧条件下孵育6小时(图1a)。用抗pgk1抗体进行的免疫荧光分析显示缺氧诱导pgk1的细胞核周围积累。因为线粒体也定位在细胞核周围区域,所以细胞与抗pgk1抗体和荧光线粒体染料mitotracker共染色。如图1a所示,pgk1与线粒体共定位,表明在缺氧刺激后pgk1易位至线粒体。这一发现得到了包括线粒体标志物coxiv和作为对照的胞质溶胶微管蛋白的细胞分级分析的支持,这显示导致hif1α积累(图9a,左图)的缺氧在u87和u251gbm细胞二者中均诱导pgk1线粒体易位(图9a,右图)。定量分析显示,约10%的胞质溶胶pgk1易位至线粒体(图9b)。因为延长的缺氧刺激增强了由hif1α介导的pgk1表达(marin-hernandez等,2009;semenza,2010),下一步将检验pgk1是否以hif1α依赖的方式易位至线粒体。通过sirna的hif1α耗竭不阻断缺氧诱导的pgk1线粒体易位,表明该过程的发生独立于hif1α(图9c)。
为了确定pgk1是否与线粒体的外膜结合或与易位至其中,使用从经缺氧刺激或未经缺氧刺激的u87和u251细胞分离的线粒体进行蛋白酶k保护测定。包括外膜标志物tom20和线粒体内标志物coxiv在内作为对照(hitosugi等,2011)。在不存在tritonx-100(其使线粒体的外膜和内膜增溶)的情况下,tom20(而非pgk1和coxiv)被蛋白酶k处理完全消化,而tritonx-100的存在使得pgk1和coxiv能进行蛋白酶k消化(图9d)。相比之下,短暂的毛地黄皂苷处理(其损伤线粒体的外膜,但不损伤内膜)对线粒体pgk1进行蛋白酶k消化的可及性的影响有限(图9e)。此外,线粒体亚分级显示pgk1与线粒体基质蛋白mnsod共分离,但不与内膜蛋白质timm22和膜间隙蛋白质细胞色素c共分离(图1b),表明pgk1易位至线粒体基质中。这些发现进一步得到免疫金透射电子显微术分析的支持,这表明缺氧诱导pgk1易位至线粒体中(图1c)。
map激酶活化在缺氧诱导的细胞活性中发挥工具性作用(kronblad等,2005;wykosky等,2011;riley等,1986)。用jnk抑制剂sp600125、p38抑制剂sb203580或mek/erk抑制剂u0126预处理u87细胞分别阻断缺氧诱导的c-jun、mapk/apk2和erk1/2的磷酸化(图10a)。免疫印迹分析显示,只有mek/erk抑制显著降低u87(图1d)和u251细胞(图10b)中缺氧诱导的pgk1线粒体易位。这些结果得到免疫荧光分析的结果的支持(图10c)。此外,flag-erk2k52r激酶失活突变体的表达阻断缺氧(图10d)和活性ha-mek1q56p突变体(图10e)诱导的pgk1线粒体易位。因此,erk1/2活化对pgk1线粒体易位是必要和充分的。与该结论一致,egf刺激(图1e)或癌基因k-rasg12v在bxpc-3人胰腺癌细胞(无内源性ras突变)中的表达以及b-rafv600e在chl1人黑素瘤细胞(无内源性b-raf突变)中的表达(flockhart等,2009;yun等,2009)(图1f)诱导pgk1线粒体易位;值得注意的是,这种易位被u0126处理或erk2k52r激酶失活突变体的表达阻断。
为了检验erk1/2是否与pgk1相互作用,用抗erk1/2抗体进行免疫沉淀的pgk1的免疫印迹分析,并显示缺氧刺激后erk1/2与pgk1缔合(图10f)。此外,用经纯化的重组活性his-erk2与gst-pgk1孵育的体外gst拉下测定显示这两种蛋白质彼此直接相互作用(图10g)。
由共同对接(commondocking,cd)结构域和谷氨酸/天冬氨酸(ed)位点组成的map激酶的对接槽(dockinggroove)用作多种map激酶相互作用分子的共同对接区域(lu和xu,2006)。cd结构域中的d316和d319以及erk2的ed位点中的t157和t158对于其底物的识别是重要的(lu和xu,2006)。免疫共沉淀测定显示内源性pgk1与flag-erk2d316/d319n和flag-erk2t157/t158e的结合大幅降低,并且完全无法与具有cd结构域和ed位点的组合突变的erk2突变体相互作用(图10h)。这些结果表明pgk1与erk2对接槽结合。
erk底物通常具有对接结构域,其特征在于一串碱性残基,随后是lxl基序(l表示亮氨酸,但也可以是异亮氨酸或缬氨酸;x表示任何氨基酸)(yang等,2012)。用scansite程序分析pgk1氨基酸序列鉴定该推定的erk结合序列74-dkyslepvave-84(seqidno:6)、170-hrahssmvgvn-180(seqidno:7)和273-aekngvkitlp-283(seqidno:8),其在v81/v83、v177/v179和v278/i280/l282分别包含lxl基序。s-flag-biotin(sfb)-pgk1蛋白的链霉亲和素拉下表明仅pgk1v278a/i280r/l282r突变显著降低其与erk1/2的结合(图10i)。这些结果表明erk2对接槽在v278/i280/l282处与pgk1中的对接结构域结合。
为了确定pgk1是否是erk1/2的底物,进行了体外激酶测定,显示经纯化和活性的erk2使经细菌纯化的pgk1磷酸化(图1g)。erk1/2使其具有p-x-s/tp(其中x可以是任何氨基酸)共有序列的底物磷酸化(ji等,2009)。pgk1氨基酸序列的分析显示pgk1仅具有一个erk1/2磷酸化基序,其在s203处。如通过放射自显影测定和使用特异性抗pgk1ps203抗体的免疫印迹分析证实(图1g),s203至ala的突变在体外用[γ32p]-atp消除erk2介导的pgk1磷酸化。免疫荧光分析显示缺氧刺激导致线粒体中磷酸化pgk1s203的积累(图11a)。此外,sfb标记的野生型(wt)pgk1和pgk1s203a被链霉亲和素琼脂糖珠拉下表明,u0126处理和pgk1s203a突变(图1h)而非sp600125处理(图11b),消除了u87细胞中缺氧诱导的pgk1s203磷酸化。与这些发现一致,erk2k52r激酶失活突变体的表达阻断活性mek1q56p诱导的pgk1s203磷酸化(图11c)。值得注意的是,pgk1s203a对缺氧诱导的线粒体易位有抗性(图1i和11d)。相比之下,磷酸化-模拟pgk1s203d突变体能够在不存在缺氧刺激下在线粒体中积累(图1i和11d),表明erk1/2介导的pgk1在s203处的磷酸化对于pgk1线粒体易位是必需的。使用抗磷酸pgk1s203抗体对具有或不具有免疫耗竭(immune-depletion)的胞质溶胶pgk1的定量显示,磷酸化pgk1的耗竭仅去除了总pgk1蛋白的约10%(图11e),这进一步支持了仅小部分的pgk1易位至线粒体的发现。
缺氧导致egfr活化(michelson等,1985)。用egfr抑制剂ag1478处理阻断了缺氧诱导的egfr磷酸化(图11f)、erk活化以及pgk1的磷酸化和线粒体易位(图11g)。这些结果表明缺氧通过活化egfr来诱导erk活化和pgk1线粒体易位。与这一发现一致,也观察到egf诱导的pgk1s203磷酸化(图11h)以及s203磷酸化依赖性pgk1线粒体易位(图11i)。此外,erkk52r的表达阻断了k-rasg12v和b-rafv600e诱导的pgk1s203磷酸化(图11j)。这些发现表明缺氧、egfr的活化以及致癌性k-ras和b-raf的表达诱导erk依赖性磷酸化和pgk1线粒体易位。
实施例2-pin1与磷酸化pgk1结合并使其顺-反异构化用于pgk1线粒体易位
erk-磷酸化的ps/tp-肽序列中的ser或thr可被肽基脯氨酸异构酶(与有丝分裂a1中的never相互作用的蛋白(proteininteractingwithneverinmitosisa1)(pin1),其催化其顺-反异构化(lu和zhou,2007;zheng等,2009))识别。pin1调节其底物的亚细胞再分布(yang等,2012)。确定erk调节的pgk1磷酸化是否导致pgk1的pin1依赖性构象变化以及随后的pgk1线粒体易位。免疫共沉淀分析显示,缺氧刺激显著提高内源性pin1与pgk1之间的相互作用,其被u0126处理阻断(图2a)。此外,缺氧刺激诱导内源性pgk1与琼脂糖珠固定化的wtgst-pin1而非gst-pin1ww结构域突变体牢固结合,其防止pin1与ps/tp底物结合(图2b)。与其wt对应物相比,pgk1s203a无法与gst-pin1相互作用(图2c)。pgk1和pin1蛋白质结构的分子建模分析显示,pgk1的s203可邻近ww结构域中的k63和r69磷酸结合口袋(phosphobindingpocket)(图12a)(michelson等,1985;yaffe等,1997),表明pin1ww结构域能够与磷酸化的s203p结合。通过体外结合测定证实erk1/2需要pin1和pgk1之间相互作用,其显示经纯化的his-pgk1仅在erk2和atp的存在下与经纯化的wtgst-pin1而非gst-pin1ww突变体结合(图2d)。此外,经纯化的pgk1s203d而非pgk1s203a能够在没有缺氧刺激的情况下(图2f)拉下u87细胞中的经纯化的gst-pin1(图2e)或内源性pin1。
为了进一步检验pgk1的磷酸化s203/p204基序是否是pin1的底物,合成了含有磷酸化或非磷酸化s203/p204的pgk1寡肽。wtgst-pin1(而非无催化活性的gst-pin1c113a突变体)使磷酸化的s203/p204肽(图2g,左图)而非其非磷酸化的对应物(图2g,右图)异构化。此外,gst-pin1使磷酸化-模拟d203/p204肽异构化,其以较磷酸化s203/p204肽更低的效率、但较非磷酸化的肽更高的效率发生(图2g,左图)。与该发现一致,his标记的磷酸化-模拟d203/p204肽以比磷酸化肽更低的效率、但以比其非磷酸化的对应物更高的亲和力与gst-pin1结合(图12b)。细胞级分分析显示,约15%的pgk1s203d易位至线粒体中(图12c),这可反映了pgk1s203d.p204被pin1异构化的效率低以及用于pgk1结合的可用pin1的量有限。这些结果表明pin1特异性地使pgk1内的磷酸化s203/p204异构化。
为了确定pin1在pgk1线粒体易位中的作用,使用缺氧来刺激具有wtpin1或pin1c113a突变体(图2h,左图)的重建表达的pin1+/+、pin1+/-或pin1-/-小鼠胚胎成纤维细胞。pin1缺乏完全阻断了缺氧诱导的pgk1线粒体易位,而该阻断通过wtpin1而非pin1c113a突变体的重新表达被挽救(图2h,右图)。此外,pin1缺乏阻断磷酸化-模拟pgk1s203d突变体的线粒体易位,并通过wtpin1而非pin1c113a突变体的重建表达挽救pgk1s203d易位的失败(图2i)。这些结果表明erk1/2介导的pgk1磷酸化导致pin1与pgk1的结合,其进而导致pgk1的顺-反异构化和随后的线粒体易位。
实施例3-pin1调节pgk1与tom复合物的结合
几乎所有的线粒体前体蛋白质(pre-protein)都是通过一般入口进入的,所述入口是外膜的移位酶(tom)。三种受体蛋白tom20、tom70和tom22作为tom复合物的一部分发挥作用。tom20充当一般入口受体,并且是具有前序列(presequence)的底物的初始识别位点(chacinska等,2009)。前序列通常位于前体蛋白质的氨基端并形成带正电荷的两亲性α螺旋,是线粒体靶向信号的经典类型(chacinska等,2009)。pgk1的结构分析显示其在n-端包含α-螺旋(第38至53位氨基酸)(图13a)。免疫共沉淀分析显示缺氧刺激导致pgk1与tom20之间的相互作用(图3a)。pin1缺乏消除了这种相互作用,其通过wtpin1而非pin1c113a突变体的重建表达(图3b)被挽救。此外,在pin1存在或不存在下,经纯化的wtgst-pgk1或gst-pgk1s203d突变体与经纯化的his-tom20的孵育显示,在pin1存在(而非不存在)下,gst-pgk1s203d而非wtgst-pgk1能够与tom20结合(图3c)。这些结果表明,pin1是磷酸化pgk1与tom复合物结合所需要的。
为了确定pgk1线粒体易位所需的pgk1的前序列,使包含α-螺旋的n-末端的1至57个氨基酸缺失,并且将该蛋白质在u87细胞中表达为c末端sfb标记的蛋白质。与其wt对应物不同,sfb-pgk1δ1-57丧失了与tom20相互作用的能力(图3d)。α螺旋中带正电荷的r39、k41和k48突变为ala,表明pgk1r39/k41a而非pgk1k48a消除了pgk1与tom20的相互作用(图3e),这有力地表明r39/k41是参与与tom复合物结合的pgk1残基。值得注意的是,如通过免疫印迹和免疫荧光分析所证明的(图3h),缺氧刺激u87细胞后,pgk1δ1-57突变体(图3f)和pgk1r39/k41a突变体(图3g至h)都不能易位至线粒体中(图3f至g)。在u251细胞中观察到pgk1r39/k41a的相似结果(图13b)。鉴于pgk1r39/k41未暴露在pgk1蛋白结构的表面(图13c),这些结果有力地表明pgk1中ps203.p204结合的pin1依赖性顺-反异构化暴露了包含pgk1的r39/k41残基(用于被tom复合物识别)的线粒体靶向信号。为了进一步支持这一结论,将经纯化的gst-pgk1s203d突变体与经纯化的pin1进行孵育或不与其进行孵育用于异构化,然后用识别38-qrikaa-43(seqidno:9)的特异性抗pgk1抗体进行免疫沉淀。用抗gst抗体的免疫印迹分析显示,针对38-qrikaa-43的抗pgk1抗体在pin1的存在下成功识别gst-pgk1s203d,但在pin1不存在下不识别(图13d)。这些结果表明,pin1调节的pgk1的异构化暴露了38-qrikaa-43残基,使得其可被肽特异性抗体识别。
实施例4-线粒体pgk1使pdhk1磷酸化
缺氧增强糖酵解途径,并且导致丙酮酸被转化成乳酸而不是用于线粒体氧化(vanderheiden等,2009;semenza,2010)。如所预期,在分离的线粒体中,缺氧降低14c标记的丙酮酸向14c标记的co2的转化速率(图4a)。为了确定线粒体pgk1是否调节pdh复合物介导的丙酮酸转化为co2的速率,用短发夹状rna(shrna)耗竭pgk1,并且在u87和u251细胞中重建rna抗(r)干扰v5标记的wtpgk1、rpgk1s203a或rpgk1r39/k41a的表达。pgk1耗竭显著抵消了缺氧诱导的丙酮酸向co2转化的抑制,并且通过在u87细胞中wtrpgk1而非rpgk1s203a或rpgk1r39/k41a的重建表达来挽救该抑制(图4a和14a)。对于用[1-14c]-丙酮酸孵育的缺氧或egf刺激的细胞也获得类似的结果(图14b)。这些结果表明线粒体pgk1调节pdh复合物的活性。
与这一发现一致,用抗pdhk1抗体对来自u87细胞的线粒体免疫沉淀的pgk1的免疫印迹分析显示缺氧诱导内源性pgk1与pdhk1的相互作用(图4b)。此外,链霉亲和素拉下测定显示,在缺氧刺激后sfb标记的pdhk1而非pdhk2、pdhk3或pdhk4与内源性pgk1相互作用(图14c)。此外,将其中经细菌纯化的sumo标记的pdhk1蛋白与经纯化并固定化的gst或gst-pgk1混合的体外结合测定显示pgk1与pdhk1直接结合(图4c)。这些结果表明缺氧导致pgk1与pdhk1之间的直接相互作用。
在糖酵解途径中,pkm2和pgk1催化仅两种生成atp的反应。pgk1催化的1,3-bpg转化为3-pg和atp是可逆反应(bernstein和hol,1998),使得pgk1也可利用atp作为磷酸供体。pkm2发挥蛋白激酶的功能(yang等,2012)。为了测试pgk1是否可充当使pdhk1磷酸化的蛋白激酶,通过将经细菌纯化的pgk1、pdhk1和atp混合来进行体外磷酸化测定。pdhk1的胰蛋白酶消化物的液相色谱耦联orbitrap质谱(lc-ms/ms)分析显示pgk1在s337或t338处使pdhk1磷酸化(图4d)。具有[γ32p]-atp的磷酸氨基酸分析显示pgk1主要在苏氨酸处使pdhk1磷酸化(图14d),表明pdhk1t338被磷酸化。此外,wtpgk1而非pgk1t378p激酶失活突变体(chiarelli等,2012)能够在[γ32p]-atp存在下使wtpdhk1而非pdhk1t338a磷酸化,其通过放射自显影术和特异性抗磷酸pdhk1t338抗体二者进行检测(图4e)。相比之下,相邻pdhk1s337a的突变对pgk1介导的pdhk1磷酸化没有影响(图14e)。这种磷酸化通过与过量的3-pg孵育而被消除(图14f),这表明pdhk1和3-pg彼此竞争被pgk1磷酸化。此外,在孵育经纯化的pgk1、pdhk1和1,3-bpg(其通过将甘油醛磷酸脱氢酶(gapdh)与甘油醛-3-磷酸和nad+孵育产生)后,pdhk1t338被磷酸化(图14g)。这些结果表明,atp和1,3-bpg二者均可以是体外pdhk1磷酸化的磷酸供体。
为了鉴定用于pgk1介导的pdhk1磷酸化的生理磷酸供体,提取线粒体并且将线粒体裂解物与经纯化的pgk1在外源性atpase存在或不存在下混合。如图14h所示,与水解线粒体atp的atpase孵育消除了pgk1介导的pdhk1t338磷酸化。这些结果有力地表明在线粒体中pgk1利用atp作为磷酸供体以使pdhk1磷酸化。鉴于用于pgk1依赖性pdhk磷酸化的atp的km(0.56±0.053mm)比u87和u251细胞中atp的生理线粒体浓度(在正常氧和缺氧条件下为2.5至3.5mm(图14j))低得多(图14i),这些结果表明在线粒体中pgk1能够利用atp高效地磷酸化pdhk1。
使用pdhk1pt338抗体从线粒体提取物中耗竭磷酸化pdhk1大幅降低了线粒体中pdhk1的量,这表明在缺氧刺激后,大部分pdhk1被线粒体中的pgk1磷酸化(图14k)。为了确定细胞中pgk1是否磷酸化pdhk1,在具有或不具有wtrpgk1或rpgk1t378p催化失活突变体的重建表达的情况下,在具有pgk1shrna的u87和u251细胞中耗竭pgk1表达(图14l)。线粒体级分分析显示rpgk1的无活性突变体和wtrpgk1二者均能够易位到线粒体中(图14m)。免疫印迹分析显示pgk1耗竭阻断缺氧诱导的pdhk1t338磷酸化,其通过wtrpgk1而非rpgk1t378p的重建表达被挽救(图14n)。此外,与其wt对应物具有相当糖酵解活性的rpgk1s203a的重建表达(图14o)在缺氧条件下无法诱导pdhk1t338磷酸化(图4f)。已知pdhk1通过fop2-成纤维细胞生长因子受体(fop2-fibroblastgrowthfactorreceptor,fgfr)1(致癌的可溶性的fgfr1融合蛋白)在酪氨酸残基处被磷酸化(hitosugi等,2011)。与先前的发现(hitosugi等,2011)一致,在缺氧刺激后,由活化的fop2-fgfr介导的pdhk1y243磷酸化没有改变(图14p)。鉴于用pdhk1pt338抗体在线粒体中耗竭磷酸化的pdhk1大幅降低了线粒体中pdhk1的量,这些结果表明在缺氧期间fgfr不参与pdhk1的调节。这些结果表明pgk1发挥蛋白激酶的作用,并在缺氧条件下使线粒体中的pdhk1t338磷酸化。
实施例5-通过pgk1的pdhk1磷酸化使pdhk1活化
为了确定pgk1是否通过磷酸化调节pdhk1活性,通过进行体外蛋白激酶测定来检验pgk1对pdhk1磷酸化的pdh的影响。在不存在或存在wtpgk1或pgk1t378p突变体和atp的情况下,将经细菌纯化的his-pdh与经纯化的wtpdhk1或pdhk1t338a混合显示通过wtpgk1而非pgk1t378p显著增强了在ser293处pdhk1依赖性pdh磷酸化(图5a)。相比之下,基础pdh磷酸化活性与wt对应物相同的pdhk1t338a未表现出pgk1增强的pdh磷酸化(图5a)。这些体外结果在u87和u251细胞中得到验证,这表明pgk1shrna的表达阻断缺氧诱导的pdh磷酸化,并且磷酸化中的该缺陷被wtrpgk1而非rpgk1s203a(图5b)、rpgk1t378p(图15a)或rpgk1r39/k41a(图15b)(具有与其wt对应物(图14n)相当的糖酵解活性)的重建表达挽救。值得注意到是,表达不易位到线粒体中的rpgk1r39/k41a的细胞用显著增强pdhk1表达的长期缺氧刺激并没有表现出增强的pdh磷酸化(图15c)。这些结果表明,pdhk1的完全活化需要pgk1依赖性磷酸化。
为了进一步检验在pdh调控下通过pgk1使pdhk1磷酸化的效果,从u87和u251细胞以及用wtrpdhk1或rpdhk1t338a重建的其表达中耗竭pdhk1(图5c,左图)。pdhk1耗竭阻断缺氧诱导的pdh磷酸化,其通过wtrpdhk1而非rpdhk1t338a的重建表达被挽救(图5c,右图)。此外,egf处理(图5d)或k-rasg12v和b-rafv600e的表达(图5e)导致增强由rpgk1s203a的重建表达(图5d)或激酶失活erkk52r的表达(图5e)阻断的pdhk1t338和pdhs293的磷酸化。这些结果表明,缺氧、egfr活化以及k-rasg12v和b-rafv600e的表达均导致pgk1介导的pdhk1磷酸化,其增强pdhk1对pdh磷酸化的活性。
实施例6-pgk1介导的pdhk1磷酸化抑制线粒体丙酮酸代谢并促进糖酵解和谷氨酰胺分解驱动的脂质合成
pdhk1磷酸化pdh并抑制其活性(holness和sugden,2003;kim等,2006)。为了确定pgk1依赖性pdhk1磷酸化在调节线粒体功能中的作用,将14c标记的丙酮酸与来自缺氧刺激的u87细胞(具有或不具有pdhk1耗竭以及wtrpdhk1或rpdhk1t338a的重建表达)分离的线粒体混合(参见图5c)。pdhk1耗竭发挥类似于rpgk1s203a表达(图4a)的作用,这显著增强了14c标记的丙酮酸向14c标记的co2的转化速率,并抵消了由缺氧诱导的抑制(图6a)。这些作用被wtrpdhk1的重建表达所消除。相比之下,rpdhk1t338a表达不能抑制缺氧诱导的pdh依赖性丙酮酸代谢(图6a)。用[1-14c]-丙酮酸孵育的缺氧或egf刺激的u87细胞也获得了类似的结果(图16a)。与这一发现一致,u87细胞(图6b)和u251细胞(图16b)的线粒体中乙酰辅酶a水平的产生被缺氧抑制,并且这种抑制通过pdhk1的耗竭消除,并通过wtrpdhk1而非rpdhk1t338a的重建表达恢复。值得注意的是,u87(图6c)和u251细胞(图16c)缺氧刺激24小时增强ros产生,其通过pdhk1耗竭进一步提高。与rpdhk1t338a的表达相比,wtrpdhk1的重建表达极大地抑制了ros产生(图6c和16c)。此外,wtrpdhk1的表达提高(其导致pdhk1t338磷酸化提高)(图16d,左图)引起在缺氧条件下对ros产生的抑制显著提高。相比之下,仅rpdhk1t338a的表达提高对缺氧诱导的ros产生的影响有限(图16d,右图),表明pdhk1t338磷酸化在pdhk1线粒体功能中的关键作用。与这一发现一致,与wtrpgk1的表达相比,rpgk1r39/k41a表达增强了ros产生(图16e)。此外,pgk1耗竭诱导更高的ros产生(图16f)和线粒体膜电位抑制(图16g)(其通过添加外源丙酮酸进一步增强)。与这些发现一致,egf处理抑制了氧消耗速率(ocr),并且通过用二氯乙酸(dca)pdhk抑制剂或pgk1s203a的重建表达处理细胞来减轻这种效应(图16h)。这些结果表明线粒体pgk1介导的pdhk1磷酸化有助于抑制pdh活性依赖性的丙酮酸代谢以及缺氧或egf诱导的线粒体ros产生和呼吸作用。
与缺氧增强糖酵解的概念一致(kim等,2006;papndreou等,2006),用短期缺氧刺激检测到提高的胞质溶胶丙酮酸水平(图6d和161)以及u87(图5d至e)和u251(图16i至j)细胞中乳酸的产生(图6e和16j),这不改变pgk1的表达水平和pdhk1的表达(图1d和4g)。值得注意的是,这种提高被pgk1(左图)或pdhk1(右图)的耗竭所阻断,其分别通过wtrpgk1或rpdhk1而非rpgk1s203a或rpdhk1t338a的重建表达而完全恢复。此外,与wtrpgk1的重建表达相比,rpgk1s203d的重建表达增强了乳酸的产生(图16k)。
egfr活化导致pgk1介导的磷酸化和pdhk1活化以及随后的pdh磷酸化。如所预期,egf刺激抑制丙酮酸向co2的转化(图16l)并提高乳酸产生(图16m);这些作用可通过pgk1或pdhk1的耗竭而减轻,这可通过wtrpgk1或wtrpdhk1而非rpgk1s203a或rpdhk1t338a的重建表达来挽救。这些结果表明线粒体pgk1介导的pdhk1磷酸化通过减弱线粒体丙酮酸代谢来促进胞质溶胶糖酵解。
糖酵解和谷氨酰胺分解通过用于脂肪酸合成的tca循环产生柠檬酸。胞质溶胶丙酮酸向乳酸转化的增强以及线粒体丙酮酸代谢的抑制可影响由糖酵解和谷氨酰胺水解引起的脂肪酸合成的动力学。与先前的报道一致,egfr活化使得用于谷氨酰胺代谢的谷氨酰胺酶活化(thangavelu等,2012),图6f显示egf处理抑制来源于d-[6-14c]葡萄糖的14c标记的脂肪酸合成,但大幅提高了l-[u-14c]谷氨酰胺来源的脂质合成。重要的是,与wtpgk1的重表达相比,通过rpgk1s203d的重建表达,谷氨酰胺分解促进的脂质合成显著增强(图6g)。这些结果有力地表明egfr活化通过抑制线粒体丙酮酸代谢并增强谷氨酰胺分解促进的脂质合成来减弱糖酵解来源的脂肪酸合成。
实施例7-线粒体pgk1依赖性pdhk1磷酸化促进细胞增殖和脑肿瘤发生并指示gbm患者的不良预后
线粒体pgk1调节的细胞代谢和ros产生可能调节细胞增殖。如所预期,在缺氧条件下培养4天的u87和u251细胞中pgk1和pdhk1的耗竭抑制增殖(图7a和17a)。wtrpgk1或rpdhk1的重建表达恢复细胞增殖。相比之下,rpgk1s203a(其维持了胞质溶胶而非线粒体功能)和rpdhk1t338a(其具有基础的但非pgk1增强的活性)的重建表达仅导致部分挽救这些对细胞有害的影响。与先前公开的结果一致(anastasiou等,2011),降低线粒体中缺氧诱导的ros产生的dtt处理部分挽救了具有wtpgk1或pgk1r39/k41a突变体的重建表达的u87细胞的缺氧诱导的生长抑制(图17b)。在正常氧条件下,表达活性egfrviii突变体的u87细胞中pgk1的耗竭抑制细胞增殖,其通过wtrpgk1而非rpgk1s203a的重建表达被挽救(图17c)。相比之下,从表达rpgk1s203d的细胞中观察到增强的细胞增殖,这也使细胞对谷氨酰胺剥夺(glutaminedeprivation)诱导的细胞增殖抑制更敏感(图17d)。这些结果表明线粒体pgk1依赖性pdhk1磷酸化在缺氧和正常氧条件下均促进细胞增殖。
为了确定pgk1在脑肿瘤发展中可能的线粒体功能,将具有或不具有耗竭的pgk1或pdhk1及其wt对应物、rpgk1s203a、rpgk1s203d、rpgk1r39/k41a或rpdhk1t338a的重建表达的u87或gsc11人原代gbm细胞经颅内注射入无胸腺裸小鼠中(图17e)。脑的解剖显示注射有u87细胞(图7b)或gsc11细胞(图17f)的所有动物中的肿瘤生长。相比之下,在分别注射有具有耗竭的pgk1或pdhk1的细胞的小鼠的脑中没有检测到肿瘤生长或检测到小得多的肿瘤。在内源性pgk1或pdhk1耗竭的细胞中,wtrpgk1或rpdhk1而非rpgk1s203a、rpgk1r39/k41a或rpdhk1t338的重建表达恢复了肿瘤生长,同时观察到rpgk1s203d增强的肿瘤生长(图7b和17f)。免疫组织化学(ihc)染色显示在具有其wt对应物的重组表达(而非具有rpgk1s203a或rpdhk1t338a的重建表达)的u87细胞中pgk1s203和pdhk1t338的强磷酸化。在具有wtrpgk1或rpdhk1的重建表达的情况下,肿瘤组织的ki67染色(图17g)和用tunel测定分析(图17h)显示迅速的细胞增殖和很少的凋亡细胞,相比之下,在具有rpgk1s203a或rpdhk1t338a重建表达的情况下,显示缓慢的细胞增殖和更多凋亡细胞。这些结果表明线粒体pgk1依赖性pdhk1磷酸化促进脑肿瘤发生。
为了确定线粒体pgk1依赖性pdhk1磷酸化调节pdh活性的发现的临床相关性,用具有抗磷酸pgk1s203、抗磷酸pdhk1t338和抗磷酸pdhs293抗体的50份人原代gbm标本(worldhealthorganizationiv级)进行ihc分析。通过使用具有特异性阻断肽进行ihc分析验证抗体特异性(kaplon等,2013)。如图6c所示,pgk1s203、pdhk1t338和pdhs293的磷酸化水平彼此相互关联。染色的定量显示这些相关是显著的(图18)。
将50名患者(全部在gbm的手术切除术后接受标准的辅助性放射治疗,随后用烷化剂(多数情况下为替莫唑胺)进行治疗)的存活时间与pgk1s203和pdhk1t338的肿瘤磷酸化水平进行比较(低:染色评分0至4;高:染色评分4.1至8)。对于其肿瘤具有低pgk1s203和pdhk1t338磷酸化水平的患者,中值存活时间分别为201.3周和192.4周;对于肿瘤具有高pgk1s203和pdhk1t338磷酸化水平的患者,中值存活时间分别为90.2周和82.9周。在cox多变量模型中,pgk1s203和pdhk1t338磷酸化的ihc评分是患者年龄(其为相关的临床协变量)调整后gbm患者存活的独立预测因子(图7d)。这些结果支持线粒体依赖性pgk1的pdhk1磷酸化在人gbm的临床行为中的作用,并揭示了erk1/2依赖性pgk1磷酸化、pgk1依赖性pdhk1磷酸化和gbm临床侵袭性之间的相关性。
实施例8-pgk1使组蛋白h2、cdc45和beclin-1磷酸化
如图19a所示,pgk1使组蛋白h3在ser10处磷酸化。此外,u87细胞中pgk1shrna的表达阻断egf诱导的组蛋白h3在ser10处的磷酸化,这对于基因转录和有丝分裂进程是重要的。
cdc45是启动dna复制所需的必要蛋白质。在[γ32p]-atp存在下,经纯化的野生型pgk1而非pgk1激酶失活(kd)突变体使经纯化的野生型cdc45而非cdc45s386a磷酸化(图19b)。
beclin-1参与自噬的启动。在[γ32p]-atp存在下,经纯化的pgk1使经纯化的野生型beclin-1磷酸化(图19c)。beclin-1s30a突变体很大程度上抵抗pgk1的磷酸化。
实施例9-pgk1在y324处的自磷酸化提高pgk1糖酵解酶活性
pgk1(在糖酵解中产生atp的糖酵解酶)发挥利用atp使其底物磷酸化的蛋白激酶的功能。除了使其他蛋白质磷酸化外,pgk1可经历自磷酸化。质谱分析将酪氨酸324(y324)鉴定为pgk1的自磷酸化位点。体外蛋白激酶测定显示由酪氨酸324向苯丙氨酸(y324f)的替换阻止自磷酸化(图20a至b)。pgk1酶活性测定显示,相对于野生型(wt)酶,pgk1y324f突变体的糖酵解酶活性严重降低,且与破坏atp结合的酶失活突变体pgk1t378p相当(图20c)。
***
根据本公开内容的教导,不需要过度实验就可以完成和实施本文中所公开和要求保护的所有方法。尽管已经以一些优选实施方案的方式描述了本发明的组合物和方法,但对于本领域技术人员来说显而易见的是可改变本文中所述的方法以及所述方法的步骤或步骤顺序而不脱离本发明的概念、精神和范围。更具体地,显而易见的是可以用化学和生理学上都相关的某些试剂替代本文中所述的试剂,同时实现相同或类似的结果。对于本领域技术人员显而易见的所有此类相似的替代和改变被视为在由所附权利要求书所限定的本发明的精神、范围和概念内。
参考文献
以下参考文献以为本文所述内容提供示例性步骤或其他补充性细节的程度具体地通过引用并入本文。
美国专利4,870,287
美国专利5,739,169
美国专利5,760,395
美国专利5,801,005
美国专利5,824,311
美国专利5,830,880
美国专利5,846,945
美国专利公开2008/004287
ahmadetal.,phosphoglyceralekinaseiasapromoterofmetastasisincoloncancer.internationaljournalofoncoiogy,intcrnationaljournalofoncology,43:586-590,2013.
aietal.,flnaandpgkiaretwopotentialmarkersforprogressioninhepatocellularcarcinoma.cellularphysiologyandbiochemistry:internationaljournalofexperimentalcellularphysiology,biochemistry,andpharmacology,27:207-216,2011.
anastasiouetal.,inhibitionofpyruvatekinasem2byrenctivcoxygenspeciescontributestoccllularantioxidantresponses,science,334:1278-1283.2011.
austin-wardandvillasecn,revistsmedicadechilc,126(7):838-845,1998.
bainetal.,theselectivilyofproteinkinaseinhibitors:afurtherupdate,biochem,j.,408:297-315,2007.
bcrnsteinandhol,crystalstructuresofsubstratesandproductsboundtothephosphoglyceratekinaseactivesiterevealthccatalyticmechanism,biochemistry,37:4429-4436.1998.
brahimi-hornetal.,nypoxiasignallingcontrolsmetabolicdemand.currentopinionincellbiology,19:223,229,2007.
brown,controlofrespirationandatpsynthesisinmammalianmitochondriaandcells,thebiochemicaljournal,284:1-13,1992.
bukowskietal.,clinicalcancerres.,4(10):2337-2347,1998.
cairnsetal.,regulationofcancercellmetabelismnaturereviews,cancer11:85-95,2011.
chacinskaetal.,importingmitochondrialproteins:machineriesandmechamisms,cell,138:628-644,2009.
chiarellietal.,moleeularinsightsonpathogenieeffectsofmutationscausingphosphoglyeeratekinasedeticieney,plosone,7:e32065,2012.
chowetal.,measurementofmapkinaseacrivationbyflowcytometryusingphospho-specificantibodiestomekanderk:potentialforpharmacodynamicmonitoringofsignaltransductioninhibitors,cytometry,46:72-78,2001.
christodoulidesetal.,microbiology,144(pt11):3027-3037,1998.
davidsonetal.,j.immunether.,21(5):389-398,1998.
daviesetal.,speciticityandmechanismofactionofsomecommonlyusedproteinkinaseinhibitors,biochem.j.,341:95-105,2000.
“designofprodrugs”,ed.h.bundgaard,elsevier,1985.
downeyetal.,chemotacticpeptide-inducedactivationofmek-2,thepredominantisoforminhumanneutrophils:inhibitionbywortmannin,j.biol.chem.,271:21005-21011,1996.
duncanetal.,discoveryandcharacterizationofanonphosphorylatedcyclicpeptideinhibitorofthepeptidylprolylisomerase,pinl,j.med.chem.,54:3854-3865,2011.
duanetal.,overexpressionofhumanphosphoglyceratekinase1(pgk1)inducesamultidrugresistancephenotype,anticancerresearch,22l1933-1941,2002.
englishandcobb,pharmtacologicalinhibitorsofmapkpathways,trendspharmacolsci.,23:40-45,2002.
fangetal.,phosphorylationofbeta-cateninbyaktpromotesbeta-catenintramseriptionalactivity,thejournalofbiologicalchemistry,282:11221-11229,2007.
floekhartetal.,nfatsignallingisanoveltargetofoncegenicbrafinmetastaticmelanoma,britishjournalofcancer,101:1448-1455,2009.
gaoetal.,c-mycsuppressionofmir-23a/benhancesmitochondrialglutnminaseexpressionandglutaminemetabolism,nature,458:762-765,2009.
gaoetal.,pyruvalekinasem2regulatesgenetranscriptionbyaetingasaproteinkinase,molecularcell,45:598-609,2012.
gatenbyandgillies,whydocancershavehighaerobicglycolysis?naturereviews,camcer,4:891-899,2004.
gomez-manzanoetal.,delta-24inereasestheexpressionandactivityoftopoisomeraseiandenhnncestheantigliomaeffectofirinotecan,clin.cuncerres.,12:556-562,2006.
hanibuchietal.,int.j,cancer,78(4):480-485,1998.
hrllstrandetal.,actaoncolodiea,37(4):347-353,1998.
hitosugietal.,tyrosinephosphorylationofmitochondrialpyruvatedehydrogenasekinase1isimportantforcancermetabolism,molecularcell,44:864-877,2011.
hockelandvaupel,tumorhypoxia;definitionsandeurrentclinical,biologic,andmoiecularaspects,journalofthenationalcancerinstiude,93:266-276,2001.
holnessandsugden,regulationofpyruvatedehydrogenasecomplexactivitybyreversiblephosphorylation,biochemicalsocietytransactions,31:1143-1151,2003.
hsiehetal.,currentprodrugdesignfordrugdiscovery,curr.pharm.des.,15:2236-2250,2009.
huiandhashimoto,infeetionimmun.,66(11):5329-5336,1998,
huttunenelal.,prodrugs-fromserendipitytorationaldesign,pharmacologicalrev.,63:750-771,2011.
hwangetal.,overexpressionandelevatedserumlevelsofphosphoglyceratekinase1inpancreaticductaladenocareinoma,proteomics,6:2259-2272,2006.
jietal.,egf-inducederkactivationpromotesck2-mediateddisassociationofalpha-cateninfrombeta-cateninandtransactivationofbeta-catenin,molecularcell,36:547-559,2009.
jiangetal.,pkm2regulateschromosomesegregationandmitosisprogressionoftumorcells,molecularcell,53:75-87,2014.
kaplonetal.,akeyroleformitochondrialgatekeeperpyruvatedehydrogenaseinoncogene-inducedsenescence,nature,498:109-112,2013.
kimetal.,hif-1-mediatedexpressionofpyruvatedehydrogenasekinase:ametabolicswitchrequiredforcellularadaptationtohypoxia,cellmeiabolism,3:177-185,2006.
kieinetal.,theeffeetsofanovelmekinhibitorpd184161onmek-erksignalingandgrowthinhumanlivercancer,neoplasia,8:1-8,2006.
koppenoletal.,ouowarburg′scontributionstocnrrentconceptsofcancermetabolism,naturereviews,cancer,11:325-337,2011.
kronbladetal.,erk1/2inhibitionincreasesaatiestrogentreatmentefficacybyinterferingwithhypoxia-induceddownregulationoferalpha:acombinationtherapypotentiallytargetinghypoxicanddormanttumorcells,oncogene,24:6835-6841,2005.
loweryetal.,proteomicscreendefinesthepolo-boxdomaininteractomeandidentifiesrock2asaplk1substrate,theembojournal,26:2262-2273,2007.
luandxu,erk1/2mapkinasesincellsurvivalandapoptosis,iubmblife,58:621-631,2006.
luandzhou,theprolylisomerasepin1:apivotalnewtwistinphosphorylationsignallinganddisease,naturereviews,molecularcellbiology,8:904-916,2007.
luetal.,activationofproteinkinasectriggersitsubiquitinationanddegradation,molecularandcellularbiology,18:839-845,1998-marin.hernandezetal.,hif-1aiphamodulatesenergymetabolismincancercellsbyinducingover-expressionofspecificglgcolyticisoforms,minireviewsinmedicinalchemistry,9:1084-1101,2009.
mattinglyetal.,thenitogen-activatedproteinkinase/extracellularsignal-regulatedkinasekinaseinhibitorpd184352(ci-1040)selectivelyinducesapoptosisinmalignantcchwannomacelllines,j.pharmacol.exp.ther.,316:456-465,2006.
michelsonetal.,structineofthehumanphosphogluceratekinasegeneandtheintron-mediatedevolutionand
morietal.,adualinhibitoragainstprolylisomerasepinlandcyclophilindiscoveredbyanovelreal-timefluorescencedetectionmethod,biochem.biophys,res.commin.,406:439-443,2011.
papandreouetal.,hif-imediatesadaptationtohypoxiabyactivelydown-regulatingmitochondrialoxygenconsumption,cellmetabolism,3:187-197,2006.
pezzatoetal.,ca2+-independenteffectsofsperminconpyruvatedehydrogenasecomplexactivityinenergizedratiivermitochondriaincubateeintheabsenceofexogenousca2+andmg2+,aminoacids,36:449-456,2009.
qinetal.,proc.natl.acad.sci.usa,95(24):14411-14416,1998.
rileyetal.,chromatinstructureofactiveandinactivehumanx-linkedphosphoglyceratekinasegene,somaticcellandmolecidargenetics.12:73-80,1986.
rinehartetal.,multicenterphaseiistudyoftheoralmeknhibitor,ci-1040.inpatientswithadvancednon-small-celljung,breast,colon,andpancreaticcancer,j.clin.oncol.,22:4456-4462,2004.
rocheandhiromasa,pyruvatedehydrogenasekinaseregulitorymechanismsandinhibitioninrreatingdoabetes,heartischemia,andcancer,cellularandmolecularlifesciences:cmls.64:830-849,2007.
semenza,hypoxia-induciblefactoriandcancerpathogenesis,iubmblife,60:591-597,2008.
semenza,hif-1:upstreamanddownstreamofcancermetabolism.curremopinioningeneticsanddevelopment.20:51-56,2010.
sheetal.,directregulationofcomplexibymitochondrialmef2disdisruptedinamousemodelofparkinsondiseaseandinhumanpatients,thejournalofclinicalinvestigation,121:930-940,2011.
tanietal.,molecularcloningandstructureofanautosomalprocessedgeneforhumanphosphoglyceratekinase,gene,35:11-18,1985.
tausetal.,universalandconfidentphosphorylationsitelocalizationusingphosphors.journalofproteomeresearch,10:5354-5362,2011.
thangaveluetal.,strueturalbasisfortheallostericinhibitorymechanismofhumankidney-typeglutaminase(kga)anditsregulationbyraf-mek-erksignalingincancercellmetabolism,proceedingsofthenationalacademyofsciencesusa,109:7705-7710,2012.
vandergeerandhunter,phospbopeptidemappingandphosphoaminoacidanalysisbyelectrophoresisandchromatographyonthin-layercelluloseplates,electrophoresis,15:544-554,1994.
vanderheidenetal.,understandingthewarburgeffect:themetabolicrequirementsofcellproliferation,science,324:1029-1033,2009.
wykoskyetal.,therapeutictargetingofepidermalgrowthfactorreceptorinhumancancer:successesandlimitations,chinesejournalofcancer,30:5-12,2011.
xiaetal.,c-jundown-regulationbyhdac3-dependenttranscriptionalrepressionpromotesosmoticstress-inducedcellapoptosis,molecularcell,25:259-232,2007.
xuetal.,recuced-amideinhibitorofpin1bindsinaconformationresemblingatwisted-amidetransitionstate,biochemistry,50:9545-9550,2011.
xuetal.,cyclohexylketoneinhibitorsofpin1dockinatrans-diaxialcyclohexaneconformation,plosone,7:e44226,2012.
yaffeetal.,sequence-specificandphosphorylation-dependentprolineisomerization:apotentialmitoticregulatorymechanism,scicnce,278:1957-1960,1997.
yanetal.over-espressionofcofilin-1andphosphoglyceratekirase1inastrocytomasinvolvedinpathogenesisofradioresistance,cnsneuroscienceandtherapeutics.18:729-736.2012.
yangetal.,erk1/2-dependentphosphorylationandnucleartranslocationofpkm2promotesthewarburgeffect,naturecellbiology,14:1295-1304,2012.
yangetal.,pkm2phosphorylateshistoneh3andpromotesgenetranscriptionandtumorigcncsis,cell,150:685-696,2012.
yoonetal,inhibitionofpik1andpin1by5′-nitro-indirubinoximesuppresseshumanlungcancercells,cancerlell.,316:97-104,2012.
yunelal.,glucosedeprivationcontributestothedevelopmentofkranpathwaymutationsintumorcells,science,325:1555-1559,2009.
zhangetal.,synthesisandstrueture-activityrelationshipsof3-cyano-4-(phenoxyanilino)quinolinesasmek(mapkk)inhibitors,bioorg.med.chem.lett.,10:2825-2828,2000.
zhangetal.,proteomicstudyrevealsthatproteinsinvolvedinmetabolicanddetoxiticationpathwaysarehighlyexpiessedinher-2/neu-positivebreastcancer,molecular&cellularproteontics:mcp,4:1686-1696,2005.
zhengetal.,fakphosphorylationbyerkprimesras-inducedtyrosinedephosphorykuionoffakmediatedbypin1andptp-pest,molecularcell,35:11-25,(2009).
ziekeretal,phosphoglyceratekinasriapromotingenzymeforperitonealdisseminationingastriccancer.internationaljournalofcancer,journalinternationalducancer,126:1513-1520,2010.
序列表
<110>得克萨斯州大学系统董事会
<120>作为用于癌症治疗和诊断之靶标的磷酸甘油酸激酶1的蛋白激酶活性
<130>utfc.p1259wo
<140>尚未指定
<141>2016-01-05
<150>us62/099,899
<151>2015-01-05
<160>10
<170>patentinversion3.5
<210>1
<211>14
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>1
hishishishishishisleugluserprogluargasnala
1510
<210>2
<211>14
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>2
hishishishishishisleugluaspprogluargasnala
1510
<210>3
<211>21
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>3
gcttctaacaccggaggtctt21
<210>4
<211>21
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>4
ggatgtctatgtcaatgatgc21
<210>5
<211>19
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>5
ccgaactagaacttgaaga19
<210>6
<211>11
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>6
asplystyrserleugluprovalalavalglu
1510
<210>7
<211>11
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>7
hisargalahissersermetvalglyvalasn
1510
<210>8
<211>11
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>8
alaglulysasnglyvallysilethrleupro
1510
<210>9
<211>6
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>9
glnargilelysalaala
15
<210>10
<211>13
<212>prt
<213>人工序列
<220>
<223>合成多肽
<400>10
leupheasntyrmettyrserthralaproargproarg
1510
- 还没有人留言评论。精彩留言会获得点赞!
- 日本血吸虫3-磷酸甘油醛脱氢酶基因、编码蛋白及应用的制作方法
- 耐高温磷酸丙糖同分异构酶基因及其编码的多肽和制备方法
- 耐高温转酮酶基因及其编码的多肽和制备方法
- 耐高温转醛酶基因及其编码的多肽和制备方法
- 一种多肽——甘油醛-3-磷酸脱氢酶-8.91和编码这种多肽的多核苷酸的制作方法
- 包含发酵基因卡盒的自养生物进行光驱动的CO<sub>2</sub>还原生成有机化合物以用作燃料或用作 ...的制作方法
- 一种新的多肽——甘油醛-3-磷酸脱氢酶 9.57 和编码这种多肽的多核苷酸的制作方法
- 一类茶树甘油醛-3-磷酸脱氢酶表达序列标签及其生物芯片的制作方法
- 通过加强辅酶循环再生速率提高Bacillus amyloliquefaciens 2,3-丁二醇产量的制作方法
- 一株产黑曲霉葡萄糖氧化酶的毕赤酵母工程菌及其应用的制作方法