米诺环素缓释制剂的制作方法
本发明涉及一种米诺环素缓释制剂。
背景技术:
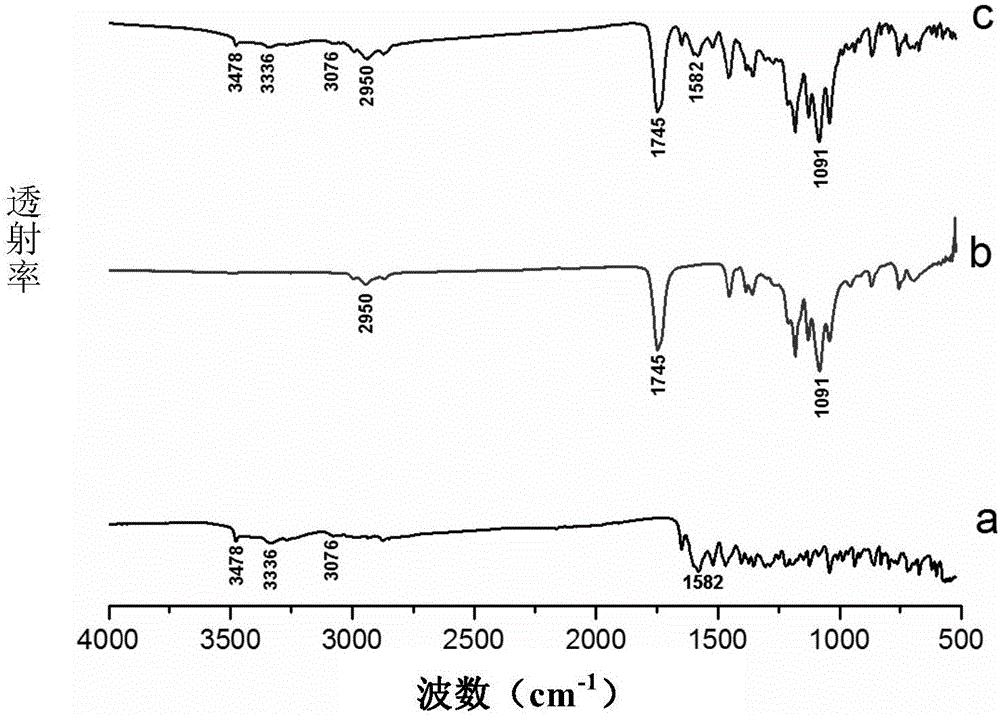
:牙周炎是口腔科常见病,是人类失牙的主要原因。对于轻度牙周炎主要为机械治疗。随着病情的进展,牙周组织破坏日益严重,牙周袋变深,单纯机械治疗很难达到满意效果,其存在于牙龈组织内或牙体组织内的致病菌很难清除完全。近年来,抗生素应用于牙周炎的治疗已越来越受到关注,局部缓释药物的治疗成为一种新型的治疗方法。盐酸米诺环素是临床上常用的牙周炎局部缓释药物,为高效、速效、长效的半合成四环素新制剂,耐药菌少、抗菌谱广、抗菌活性强,易渗透,具有独特的药物释放特点,是四环素族抗生素中抗菌活性最强且血药有效时间最长的一种。对各类牙周炎的致病菌均高度敏感,且能抑制胶原活性,促进组织愈合,具有广阔的应用与发展空间。王银甫[1]通过研究证实,盐酸米诺环素软膏遇水变硬后形成带有网孔样的被膜,有利于更好地在牙周袋内缓慢释放,且可保持局部高浓度稳定达7天,同时还可使牙槽骨的破坏速度大幅减慢,以达到长效治疗目的。聚己内酯(PCL)、聚丙交酯(PLLA)是常见的生物可降解聚酯,具备良好的机械性能、可共混性、生物相容性和降解性。聚乙二醇单甲醚(mPEG)作为良好的亲水性聚合物,其生物相容性好,无毒,免疫原性低,可通过肾脏排出体外。许亮亮等[2]采用沉淀法合成了两亲性聚己内酯-乙二醇-丙交酯(PCELA)三嵌段共聚物,但该文献中,制备得到的是共聚物的纳米胶束,而不是生物薄膜,而且没有涉及将共聚物用于缓释制剂,更没有涉及体外释放性、成膜性等的研究。陈红丽[3]等使用PEG6000合成了聚己内酯/聚氧乙烯/聚丙交酯三元共聚物,共聚物的不同单体的比例为14/14/72,25/25/50,该文献也没有涉及将共聚物用于缓释制剂,而且该文献只研究了共聚物的生物降解性,没有涉及共聚物的成膜性、口腔中的释放性等的研究。ZhengZhang[4]等合成了三嵌段共聚物(聚己内酯-丙交酯)-聚乙二醇-(聚己内酯-丙交酯),即PCLA-PEG-PCLA,该文献研究了该聚合物的生物安全性能、降解性能和预防术后粘连的屏障作用,没有涉及共聚物的成膜性、载药性及缓释性。现有技术文献[1]王银甫.盐酸米诺环素软膏和替硝唑局部用药治疗牙周炎的疗效对比分析[J].中国医药科学,2011,1(24):87-88.[2]许亮亮,陈强,李利,等.聚己内酯/聚乙二醇/聚丙交酯两亲性共聚物纳米胶束的制备与表征[J].材料导报,2006,20(11):131-133.[3]陈红丽,贝建中,王身国.生物降解高分子-聚己内酯/聚氧乙烯/聚丙交酯三元共聚物水解行为的研究[J].高分子学报,2000,(5):626-631.[4]ZhengZhang,JianNi,LiangChen,etal.BiodegradableandthermoreversiblePCLA-PEG-PCLAhydrogelasabarrierforpreventionofpost-operativeadhesion.Biomaterials,2011,32(21):4725-4736.技术实现要素:发明要解决的技术问题本发明旨在开发一种具有良好的缓释作用的米诺环素缓释制剂。用于解决上述技术问题的方案发明人发现,以聚乙二醇单甲醚(mPEG)为引发剂,左旋丙交酯(L-LA)和己内酯(ε-CL)为单体开环聚合制备的mPEG-P(LLA-co-CL)(聚乙二醇单甲醚-聚丙交酯-聚己内酯)无规共聚物,当该聚合物的数均分子量控制在8.0×104~9.0×104的范围内,L-LA和ε-CL的重复单元的比例为3比1时,可得到具有一定亲水性和良好的力学强度、同时兼具良好的载药性能和药物释放性能的可降解生物薄膜,由该可降解生物薄膜可以制得具有良好的缓释作用的米诺环素缓释制剂。具体而言,本发明包括以下内容。1.一种米诺环素缓释制剂,其特征在于,由可降解生物薄膜与米诺环素形成,所述可降解生物薄膜是以聚乙二醇单甲醚为引发剂、以左旋丙交酯和ε-己内酯为单体开环聚合而得的聚乙二醇-聚丙交酯-聚己内酯无规共聚物,所述无规共聚物的数均分子量在8.0×104~9.0×104的范围内,且具有如下(1)所示的结构式:其中,左旋丙交酯单元和ε-己内酯单元的比例为3比1。2.如上述(1)所述的米诺环素缓释制剂,所述米诺环素以米诺环素的药学上可接受的盐的形式提供。3.如上述(2)所述的米诺环素缓释制剂,所述米诺环素的药学上可接受的盐的形式是盐酸米诺环素。4.如上述(1)-(3)中任一项所述的米诺环素缓释制剂,其特征在于,所述引发剂聚乙二醇单甲醚为mPEG4000。5.如上述(1)-(3)中任一项所述的米诺环素缓释制剂,其特征在于,所述共聚物的分子量分布为1.5。6.如上述(1)-(3)中任一项所述的米诺环素缓释制剂,其特征在于,所述共聚物的数均分子量为8.1×104。7.如上述(1)-(3)中任一项所述的米诺环素缓释制剂,其特征在于,载药量为5wt%-10wt%。8.一种米诺环素缓释制剂的制备方法,其特征在于,称取可降解生物薄膜与米诺环素的药学上可接受的盐,分别溶于氯仿和甲醇中,充分混匀后静置,待气泡除尽后铺膜于聚四氟乙烯板上,室温干燥后脱膜,所述可降解生物薄膜是以聚乙二醇单甲醚为引发剂、以左旋丙交酯和ε-己内酯为单体开环聚合而得的聚乙二醇-聚丙交酯-聚己内酯无规共聚物,所述无规共聚物的数均分子量在8.0×104~9.0×104的范围内,且具有如下(1)所示的结构式:其中,左旋丙交酯单元和ε-己内酯单元的比例为3比1。9.如上述(8)所述的米诺环素缓释制剂的制备方法,其特征在于,所述米诺环素的药学上可接受的盐为盐酸米诺环素。10.如上述(8)或(9)所述的米诺环素缓释制剂的制备方法,其特征在于,所述共聚物的数均分子量为8.1×104。发明效果通过本发明,可以提供一种具有良好的缓释效果的米诺环素缓释制剂,所述米诺环素缓释制剂具有可生物降解的薄膜,该种薄膜具有一定亲水性和良好的力学强度、同时兼具良好的载药性能和药物释放性能。附图说明图1是mPEG-P(LLA-co-CL)的1HNMR图谱图2是盐酸米诺环素、聚合物和载药膜的红外光谱图a表示盐酸米诺环素,b表示聚合物,c表示载药膜图3是不同倍数下载药膜表面的扫描电子显微镜(SEM)图A:54倍,B:1000倍,C:2000倍,D:4000倍图4是不同倍数下拉伸后的载药膜表面的扫描电镜(SEM)图A:54倍,B:1000倍,C:2000倍,D:4000倍图5是共聚物1制得的盐酸米诺环素缓释制剂的抑菌圈实验结果A表示对致龋菌变形链球菌(S.m)的抑菌环,B表示对具核梭杆菌(F.n)的抑菌环图6是载药膜的激光共聚焦实验结果图A、B表示载药膜对具核梭杆菌生物膜的抗菌作用(A:0wt%,B:10wt%),C、D表示0wt%载药膜对变形链球菌的抗菌作用(C:浮游菌,D:生物膜),E、F表示10wt%载药膜对变形链球菌的抗菌作用(E:浮游菌,F:生物膜)。具体实施方式以下,参照附图详细说明本发明的具体实施方式。另外,本发明的实施方式并不限定于下述实施方式,本领域技术人员可以在本发明的技术构思范围内进行各种改变和替换。实施例一、mPEG-P(LLA-co-CL)聚合物的制备1材料与设备1.1实验材料1.2主要设备仪器名称型号生产厂家真空干燥箱XMTD-8222精宏有限公司油泵ZXZ-4型上海德英真空照明有限公司核磁分析仪Bruker瑞士Bruker凝胶渗透色谱仪Waters1515美国Waters2实验方法2.1实验原料的前处理(1)ε-己内酯:将ε-己内酯倒进圆底烧瓶中,称取少许的氢化钙(CaH2)倒入其中,搅拌反应72小时,除去微量水分,减压蒸馏得到干燥的ε-己内酯,密封干燥保存。(2)甲苯:将一定量的钠丝加入到待干燥的甲苯中,250℃下回流7-8个小时,蒸馏得到干燥的甲苯,干燥保存。(3)L-丙交酯:用天平称量左旋丙交酯置于250ml的反应球瓶内,倒入适量体积的干燥的甲苯,在105-110℃下搅拌,在丙交酯单体在甲苯中溶解完全后,快速倾倒在烧杯中,用保鲜膜封好,置于冰箱中重结晶,待晶体完全析出,使用真空泵抽滤。此类重结晶重复两次。将处理两次的L-丙交酯盛入圆底烧瓶中,加入适量的干燥甲苯,在130℃下搅拌回流,待甲苯从油水分离器中回流两个小时。将溶液倒入烧杯,用保鲜膜封好,置于冰箱中重结晶,待晶体刚刚析出,使用真空泵抽滤。此类重结晶重复两次,最后一次抽滤完成使用无水乙醚淋洗,用水泵将产物抽气三小时,得到纯化的左旋乳酸单体,将其密封后储存在干燥的容器内以备后续聚合实验用。(4)mPEG4000:将mPEG4000盛入圆底烧瓶中,加入适量的干燥甲苯,在130℃下搅拌回流,待甲苯从油水分离器中回流两个小时后减压蒸馏除去甲苯,得到干燥的mPEG4000。本发明中,mPEG的分子量太大的话,在聚合物端基起的作用大,在体内不能被代谢,且降解速度会变慢,因此优选使用mPEG4000。2.2mPEG-P(LLA-co-CL)共聚物的制备在氩气的保护下,将经前述预处理后的ε-己内酯和L-丙交酯两种单体以及引发剂mPEG迅速加入到预先烤过的反应茄形瓶中,置于45℃左右的油浴抽真空。在氩气的保护下,滴加催化剂异辛酸亚锡甲苯溶液至反应体系中,换气。将聚合瓶放在温度为120~150℃的二甲基硅油的油浴锅中进行聚合。反应结束后,待体系冷却至室温,加入适量的二氯甲烷溶解,逐滴滴加反应产物溶液至搅拌的石油醚中,进行沉降。将沉降后的聚合产物放在常温下的真空干燥箱内至恒重。本发明中,反应温度优选控制在120~150℃,如果低于120℃,ε-己内酯单体的反应活性较低,制备的共聚物中ε-己内酯的比例往往低于设计比例,聚合物也趋于呈现嵌段的特性。温度超过150℃,即较高的反应温度下,异辛酸亚锡的催化活性大大降低,不利于反应的进行。140℃时是最为理想的反应温度。制备例1在氩气的保护下,将经前述预处理后的2.2278gε-己内酯和4.2222gL-丙交酯两种单体以及引发剂0.3gmPEG4000一起迅速加入到预先烤过三次的反应茄形瓶中,置于45℃的油浴抽真空4h。在氩气的保护下,通过微量注射器滴加1g/mol的异辛酸亚锡甲苯溶液32.25μl(单体总质量的千分之0.5)至反应体系中,换气三次。将聚合瓶放在温度为140℃的二甲基硅油的油浴锅中进行聚合24h。反应结束后,待体系冷却至室温,加入适量的二氯甲烷溶解,逐滴滴加反应产物溶液至搅拌的石油醚中,进行沉降。将沉降后的聚合产物放在常温下的真空干燥箱内至恒重,得到mPEG-P(LLA-co-CL)共聚物1。制备例2在氩气的保护下,将经前述预处理后的2.2304gε-己内酯和4.2298gL-丙交酯两种单体以及引发剂0.3gmPEG4000一起迅速加入到预先烤过三次的反应茄形瓶中,置于45℃的油浴抽真空4h。在氩气的保护下,通过微量注射器滴加1g/mol的异辛酸亚锡甲苯溶液32.22μl(单体总质量的千分之0.5)至反应体系中,换气三次。将聚合瓶放在温度为120℃的二甲基硅油的油浴锅中进行聚合一天一夜。反应结束后,待体系冷却至室温,加入适量的二氯甲烷溶解,逐滴滴加反应产物溶液至搅拌的石油醚中,进行沉降。将沉降后的聚合产物放在常温下的真空干燥箱内至恒重,得到mPEG-P(LLA-co-CL)共聚物2。制备例3在氩气的保护下,将经前述预处理后的2.2232gε-己内酯和4.2152gL-丙交酯两种单体以及引发剂0.3gmPEG4000一起迅速加入到预先烤过三次的反应茄形瓶中,置于45℃的油浴抽真空4h。在氩气的保护下,通过微量注射器滴加1g/mol的异辛酸亚锡甲苯溶液32.28μl(单体总质量的千分之0.5)至反应体系中,换气三次。将聚合瓶放在温度为150℃的二甲基硅油的油浴锅中进行聚合一天一夜。反应结束后,待体系冷却至室温,加入适量的二氯甲烷溶解,逐滴滴加反应产物溶液至搅拌的石油醚中,进行沉降。将沉降后的聚合产物放在常温下的真空干燥箱内至恒重,得到mPEG-P(LLA-co-CL)共聚物3。2.3聚合物的表征2.3.1核磁共振波谱分析(NMR)1HNMR用来表征制备的mPEG-P(LLA-co-CL)共聚物的单体组成比例以及序列结构分布。具体操作如下:称量约5mg的mPEG-P(LLA-co-CL)共聚物,溶于CDCl3中,采用德国的BrukerAV400MHz核磁共振波谱分析仪测试mPEG-P(LLA-co-CL)的1HNMR。在核磁氢谱图中得到mPEG-P(LLA-co-CL)共聚物中分子上质子氢的出峰位置及相对积分峰面积,从而可以确定mPEG-P(LLA-co-CL)共聚物的结构。以下,以mPEG-P(LLA-co-CL)共聚物1为例,详述共聚物的结构解析过程。图1是mPEG-P(LLA-co-CL)共聚物1的1HNMR图谱,所有质子的化学出峰位置已经一一标注在其中。从1HNMR图谱可以看出,mPEG-P(LLA-co-CL)共聚物1中,L-LA单元中的质子出峰为g和h,ε-己内酯结构单元中各质子的出峰标注为b、c、d、e、f,mPEG的出峰位置为a。两种单体的出峰具体如下:己内酯单元中所有质子在核磁氢谱上的化学位移出峰位置分别为1.35ppm、1.65ppm、2.31-2.39ppm和4.06-4.14ppm,分别对应于γ亚甲基上的质子(d)、β和δ亚甲基上的质子(e和c)、α亚甲基上的质子(b)以及ε亚甲基上的质子(f)。左旋丙交酯重复单元中所有氢质子在核磁谱图上的化学出峰位置分别在1.49-1.55ppm和5.05-5.18ppm之间,与其相对应的是甲基上的氢质子(h)和次甲基上的氢质子(g)。b、c、d、e、f、g、h为CL和LA相互影响下所产生的各类偏峰。由上述核磁共振波谱分析可知,mPEG-P(LLA-co-CL)共聚物1的结构如下式(1)所示:其中,L-LA和ε-CL重复单元的比例为3:1。用与上述2.3.1项同样的方法,对由制备例2制得的共聚物2和由制备例3制得的共聚物3进行了表征,结果显示:共聚物2、3也具有式(1)所示的结构。2.3.2凝胶渗透色谱仪(GPC)mPEG-P(LLA-co-CL)共聚物的数均分子量(Mn)及其分子量分布(PDI)是采用美国沃特斯公司的型号为water1515的凝胶色谱分析仪,以THF作为洗脱溶剂,1mL/min的流速进行测定。通过以上测定,mPEG-P(LLA-co-CL)共聚物的数均分子量和分子量分布如表1所示。表1:共聚物1共聚物2共聚物3数均分子量(Mn)806788100481596分子量分布(PDI)1.51.51.5这里,mPEG-P(LLA-co-CL)共聚物中丙交酯和己内酯重复单元的比例优选为3:1,且共聚物的数均分子量优选在8万~9万的范围内,发明人意外地发现,当满足上述条件时,由该共聚物膜制得的盐酸米诺环素口腔缓释制剂能够同时具备长效的缓释性能与良好的力学性能(成膜性),而这两种性能对于口腔缓释制剂尤为重要。二、盐酸米诺环素制剂的制备1实验部分1.1材料与设备1.1.1实验材料盐酸米诺环素,黄色结晶性粉末,无臭、味苦,纯度99%(玛雅试剂有限公司);甲醇,无色透明液体,上海泰坦科技有限公司;三氯甲烷,无色透明液体,国药集团化学试剂有限公司;脑心浸液(BHI),美国BD公司;琼脂粉(批号:110914),上海伯奥生物科技有限公司;柠檬酸,汕头市光华化学厂;十二水合磷酸氢二钠,国药集团化学试剂有限公司;pH精密试纸(6.4~8.0),国药集团化学试剂有限公司;国际标准菌株变形链球菌(UA159),具核梭杆菌(AT25586),上海市口腔医学重点实验室微生物实验室提供。1.1.2主要设备JA2003N电子天平(上海精密科学仪器有限公司);磁力搅拌器(B11-3,上海司乐仪器有限公司);pH测试仪(上海精密科学仪器有限公司);游标卡尺(精度0.02mm,上海量具刃具厂);恒温培养箱(SANYO,日本三洋机电公司,日本);厌氧培养箱(WhitleyDG250,Donwhitleyscientific公司,英国);分光光度计(UV-1601,Shimadzu公司,日本)。1.2实验方法1.2.1载药膜的制备精密称取1.5gmPEG-P(LLA-co-CL)和适量原料药盐酸米诺环素,分别溶于15mL氯仿和1mL甲醇中,然后将盐酸米诺环素溶液缓慢滴加入聚合物溶液中,充分混匀后静置,待气泡除尽后铺膜于聚四氟乙烯板上,室温干燥72小时后脱膜,制得含药量分别为5wt%、8wt%和10wt%的三种载药膜及不含药的空白膜。用打孔器将膜片打成6mm直径大小的圆形膜片,紫外灯照射30min灭菌,密封于洁净棕色瓶内备用。每片药膜含药物0.5~1.0mg。1.2.2SEM表征薄膜的表面形貌每组膜随机选取一个裁剪成3mm×10mm大小的小长条形,喷金处理后在扫描电镜上对每组薄膜的表面形貌进行分析。1.2.3载药膜的理化性能测试1.2.3.1质量均匀性测定随机选取每组膜中的一个膜,在膜的5处不同位置裁剪出1cm×1cm的膜片,分别用电子天平称量,计算平均值。1.2.3.2厚度均匀性测定随机选取每组膜中的一个膜,对膜的5个不同位置的厚度用螺旋测微器进行测量,计算平均值。1.2.3.3表面PH值测定将各组膜剪切成相同大小1cm×1cm的膜片,每组平行样5个,置于相应编号的培养皿中,注入McIlvaine缓冲液(pH6.6),浸泡2小时后,用PH试纸进行测定。1.2.3.4机械性能测定按照GB/T1040.3-2006《塑料拉伸性能的测定第3部分:薄膜和薄片的试验条件》,采用标准模具将薄膜裁剪成宽4mm,标距25mm的哑铃型样品,每组平行样5个。样品裁成后,常温常压存放12小时,释放裁剪过程中造成的应力。拉伸速率200mm/min,测试温度(25±2)℃,取平均值。1.2.4载药膜的药物缓释性能研究1.2.4.1McIlvaine缓冲液的配制精密称取十二水磷酸氢二钠35.814g,无水柠檬酸9.6065g,分别用500ml去离子水将其溶解,配得0.2mol/LNa2HPO4和0.1mol/L柠檬酸溶液后,再分别取145.5ml0.2mol/LNa2HPO4和54.5ml0.1mol/L柠檬酸混合制得pH值为6.6的McIlvaine缓冲溶液。1.2.4.2检测波长的确定配制0.02mg/ml的盐酸米诺环素溶液,在200~400nm波长范围内进行紫外吸收波长扫描,盐酸米诺环素的最大吸收波长为345nm。1.2.4.3干扰实验配制10mg/mlmPEG-P(LLA-co-CL)溶液,在200~400nm波长范围内进行紫外吸收波长扫描,发现在345nm波长处无吸收,因而可排除药膜辅料在该波长处对盐酸米诺环素药物检测的干扰。1.2.4.4标准曲线的建立精密称取盐酸米诺环素10mg溶解于50ml量瓶中,定容后,分别取100ul、250ul、375ul、500ul、750ul、1000ul、1250ul和1500ul于10ml量瓶中定容,得浓度分别为2.0ug/ml、5.0ug/ml、7.5ug/ml、10.0ug/ml、15ug/ml、20ug/ml、25ug/ml和30ug/ml的盐酸米诺环素稀释液,在345nm处测吸收度,以浓度C对吸收度A作图,并进行线性回归。1.2.4.5药物缓释实验精密称取载药膜片(药含量分别为0.5mg,0.8mg,1.0mg)放入200ml锥形瓶中,加入50ml的Mcllvaine缓冲液,置入37℃全温箱中静置。在特定时间点0.5h,1h,2h,4h,8h,12h,24h,48h,72h,96h,120h,144h,168h,192h,216h,240h,264h,288h,312h和336h用移液枪从锥形瓶中准确量取5mlMcIlvaine缓冲液,同时加入5ml温度为37℃的新鲜McIlvaine缓冲液,保持总体积不变,用紫外分光光度计测量药物的吸光度,计算累积释药量百分率。每组进行3次平行实验,实验结果取平均值。1.2.5载药膜的体外抗菌性能研究1.2.5.1菌液的配制实验选用牙周致病菌具核梭杆菌(F.nucleatum),致龋菌变形链球菌(S.mutans),用无菌接种环挑取BHI琼脂培养板表面单克隆菌落于3mlBHI液体培养基中,S.mutans于普通培养箱,F.nucleatum于厌氧培养箱37℃培养24h。染色镜检为纯培养物,分光光度计将菌液浓度调至1×106CFU/ml备用。1.2.5.2抑菌圈实验在超净工作台中,将消毒好的的BHI琼脂固体培养基倒入无菌培养皿中,冷却得到培养基平板后,用移液枪吸取50ul菌液滴加至培养基平板上,无菌L棒推匀,制得含菌培养基平板。将消毒好的圆形膜片等距离放入含菌培养基平板中,贴放好后,用无菌镊子轻压膜片使其与琼脂表面适当接触。以mPEG-P(LLA-co-CL)空白膜为对照,平行做3个培养皿。分别倒置于37℃厌氧培养箱中,24h后观察结果。用游标卡尺测量抑菌圈直径。重复实验3次,取平均值。1.2.6载药膜的体外细胞毒性研究1.2.6.1浸提液制备及分组将0wt%~10wt%MH/mPEG-P(LLA-co-CL)药膜紫外光照2h消毒后按试件表面积和培养液之比为6cm2/ml(ISO10993-12),将药膜放入含10mlDMEM细胞培养液的无菌离心管中,置于37℃恒温摇床放置24±2h。培养后细胞浸提液用直径为0.22um的滤器过滤,然后稀释分别配成50%、100%的材料浸提液。实验设置阳性对照和阴性对照,添加二甲亚砜组为阳性对照组,添加细胞培养液组为阴性对照组。1.2.6.2细胞毒性测试(MTT)采用体外细胞培养的方法,根据ISO10993-5,实验采用灵敏度高的噻唑蓝比色法(MTT法)进行体外细胞毒性检测。实验选用牙周膜成纤维细胞,细胞取自三名志愿者,年龄在20-25岁之间,拔出的第三磨牙的正常牙周组织,在DMEM细胞培养液中培养传代至第三代,配制成浓度为4×103/ml细胞悬液,然后接种于96孔板中,每孔200ul,每组6孔。37℃恒温培养24h后,吸弃培养液,将50%、100%两种浓度材料浸提液分别加入96孔板中,每孔200ul。于37℃细胞恒温箱中培养24h,每孔加入20ul(5mg/mL)MTT溶液避光培养4h。吸弃各孔液体,每孔加入200ul二甲基亚砜,室温下震荡10min,于酶联免疫仪上测得各孔在490nm处的吸光度值(OD),实验重复三次。计算各组中细胞的相对活性(RGR)。1.2.7载药膜的激光共聚焦实验将2.4×2.4cm2盖玻片置入6孔板中,每孔分别加入约2ml105CFU/ml的变形链球菌和具核梭杆菌菌液,厌氧培养24h后形成生物膜,用生理盐水冲洗2遍后,分别用不同药物含量的载药膜浸提液进行处理30min,空白膜浸提液处理组作为对照组。同时,原6孔板中的浮游菌用等量体积的药膜浸提液进行处理。30min后,生物膜样本用生理盐水轻轻冲洗5遍,同时浮游菌收集混匀,避光,分别加入LIVE/BacLightTMBacterialViabilityKit细菌存活试剂盒荧光染液,15min后将盖玻片置于激光共聚焦显微镜下观察。488nm/561nm光源激发后,活菌与SYTO9结合发出绿色荧光,死菌细胞核DNA与PI结合发出红色荧光。实验组各含三个样本。2结果2.1外观制备例1~3所制得的共聚物膜为黄色片状型薄膜,光洁完整,厚度一致,色泽均匀,无明显气泡。2.2膜表面微观分析图2为盐酸米诺环素、聚合物和载药膜的红外光谱图,其中曲线a为盐酸米诺环素,曲线b为mPEG-P(LLA-co-CL)聚合物,曲线c为载药膜,红外光谱图显示,主药与聚合物间无相互作用,因此可以推测,药物和聚合物是兼容的,可以制备成膜。图3为不同倍数下10wt%载药共聚物膜表面的扫描电子显微镜(SEM)图,其中A、B、C、D分别为54倍、1000倍、2000倍、4000倍镜下的载药膜表面微观图,可见,药物结晶颗粒自组装呈圆片状有序镶嵌在聚合物薄膜中。2.3膜的质量、厚度及表面pH分析药膜的质量和厚度均匀性是影响膜药物剂量的直接因素。结果显示,本发明的共聚物药膜的质量和厚度都较均匀,且可以根据具体需要通过溶液铸膜法来获得。载药膜表面pH值范围在6.7~7之间,与龈沟液的pH值6.6接近,偏中性,结果如表2所示。表2:不同膜的物理化学性能2.4载药膜的力学性能评价对载药膜的力学性能做了测试(表3),其中,载药膜的断裂伸长率很高、最大负荷较小、拉伸强度较小,即材料的韧性很好,较为柔软,作为牙周袋内的载药薄膜,可以很好的贴合患处。同时,对拉伸后的载药膜拍摄电镜(图4)可以看出,白色颗粒部分确实为盐酸米诺环素颗粒,黑色部分为拉伸测试中,颗粒出来后在聚合物上留下的孔洞,其中A、B、C、D分别为54倍、1000倍、2000倍、4000倍镜下拉伸后的载药膜表面微观图。表3.盐酸米诺环素药物膜的力学性能药物含量最大负荷(N)拉伸强度(MPa)断裂伸长率(%)弹性模量(N/mm)5wt%4.987.00906.331.108wt%4.227.27972.931.0110wt%4.107.12977.661.052.5载药膜的缓释性能评价2.5.1盐酸米诺环素标准曲线以盐酸米诺环素浓度C为横坐标,345nm处的吸光度A为纵坐标得回归方程:A=0.0274C+0.0014(r2=0.9999),在345nm处,盐酸米诺环素的浓度C在2.0~24.0ug/ml范围内与吸光度A有良好的线性关系。2.5.2体外缓释实验各时间点测得的吸光度按回归方程测定释药量,计算累积释药量百分率,以累积释药量百分率对时间进行作图。表4为由共聚物1制得的不同比例盐酸米诺环素药物膜体外缓释结果。表4显示,药物释放时间随着盐酸米诺环素含量的增加而缩短,10wt%盐酸米诺环素药膜缓释时间为两周,且随着药物含量的增加,药物累积释放率也明显增加。表4.盐酸米诺环素药物膜体外释放结果(n=3,pH6.6)2.6载药膜的抗菌性能评价2.6.1抑菌环实验对由共聚物1制得的盐酸米诺环素缓释制剂的抗菌性能进行了评价。盐酸米诺环素缓释制剂的抗菌试验显示,各培养基平板细菌生长良好,载药膜对S.m和F.n均有抑菌环产生。抗菌效果随着药物含量的增加而逐渐增强,且药膜对F.n的抑制作用强于对S.m的抑制作用。结果见图5,其中,A为对致龋菌变形链球菌(S.m)的抑菌环,B为对具核梭杆菌(F.n)的抑菌环。2.6.2激光共聚焦实验在空白膜浸提液组(图6A、C、D),变形链球菌、具核梭杆菌的浮游菌和生物膜的激光共聚焦图片中,绿色荧光几乎充满了整个视野,而在不同药物含量的载药膜浸提液处理组中(图6B、E、F),绿色和红色荧光各占一定比例并有部分交叠成橘黄色,即活死菌的重叠,随着药物含量的增加,生物膜和浮游菌对比对照组,死菌均居多,说明盐酸米诺环素药膜对变形链球菌和具核梭杆菌的生物膜及浮游菌均有很强的抑制作用。2.7载药膜的体外细胞毒性评价对材料的细胞毒性进行分级评定(五级)。细胞毒性0级:RGR≥100%,细胞毒性1级:75%≤RGR≤99%,细胞毒性2级:50%≤RGR≤74%,细胞毒性3级:5%≤RGR≤49%,细胞毒性4级:1%≤RGR≤24%,细胞毒性5级:RGR=0。结果评价:若实验结果为0或1级,表示属于合格材料;实验结果为2级,要结合细胞具体形态进行综合评价;实验结果为3-5级,则材料为不合格。结果见表5,6。表5.100%和50%浓度时MH/mPEG-P(LLA-co-CL)各组药膜浸提液的OD值表6.MH/mPEG-P(LLA-co-CL)各组细胞相对增值率及毒性级别由表5,6可见,除了100%浓度的10wt%MH/mPEG-P(LLA-co-CL)细胞浸提液细胞相对增值率87.45%,细胞毒性级别为1级,其他50%和100%的细胞浸提液细胞相对增值率均大于100%,细胞毒性级别均为0级。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1