一种抗肿瘤药物组合物的制作方法
本发明属于化学医药领域,一种抗肿瘤药物组合物。
背景技术:
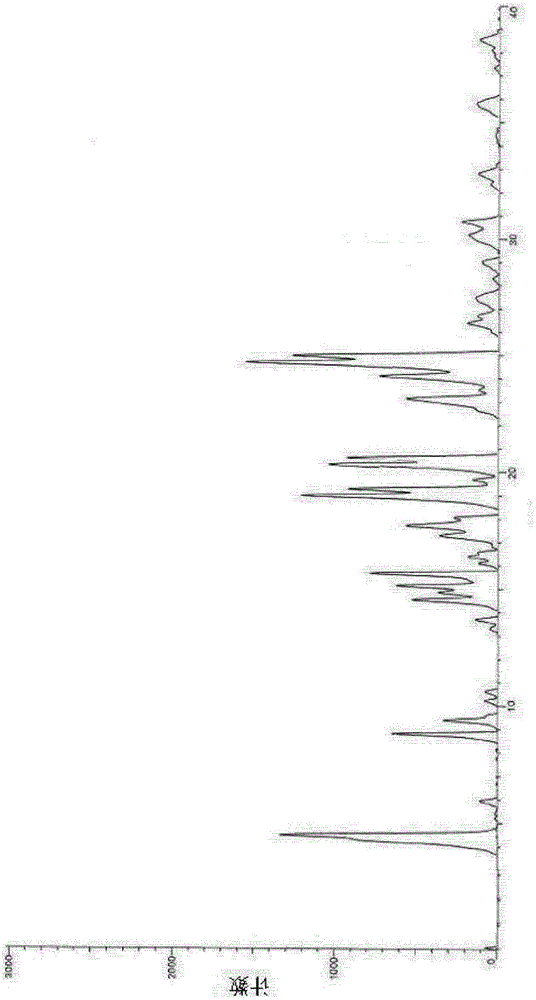
:布鲁顿氨酸激酶(bruton'styrosinekinase,btk)属于tec家族的成员。它由独特的n-端结构域即ph(pleckstrinhomology)结构域、th(techomology)同源区、sh3(srchomology3)结构域、sh2(srchomology2)结构域和催化结构域,也称sh1/tk(srchomologyl/tyrosinekinase)结构域或者激酶结构域组成(akinleyeetal:ibrutinibandnovelbtkinhibitorsinclinicaldevelopment.journalofhematology&oncology2013,6:59)。在b淋巴细胞正常发育过程中,btk基因不同蛋白区域的正确表达在b细胞的功能及多种转导途径中具有关键性作用。基于btk信号传导通路开发小分子靶向药物为b细胞类肿瘤如白血病、发性骨髓瘤及b细胞类免疫疾病的治疗提供一条全新的途径。btk在自身免疫疾病中的作用的证据已经由btk-缺失型小鼠和btk-充足型小鼠模型试验提供(killp,etal:bruton'styrosinekinasemediatedsignalingenhancesleukemogenesisinamousemodelforchroniclymphocyticleukemia.amjbloodres2013,3(1):71-83.)。在慢性淋巴细胞白血病(cll)小鼠模型中,btk-缺失型小鼠完全废止慢性淋巴细胞白血病,btk过度表达会加速白血病发病,增加死亡率。目前,数种btk抑制剂得到了开发,然而,疗效不佳。本领域知道,药物晶型的不同,会造成各种理化性质的差异,如溶解度、溶出速率、熔点、密度、硬度、光电学性质、蒸汽压力等。这些差异可反映在热力学稳定性上,如稳定型、亚稳定型和不稳定型;也可反映在物理化学稳定性上,如吸湿性、晶型转变、样品降解等。这些差异直接影响药物的处方制剂工艺、储存方法、体内药代动表现,进而影响到药物的安全性和有效性。因此,研究[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐的多晶型现象并选择出在临床治疗上有意义且稳定可控的晶型具有着十分重要的意义。此外,尽管抗癌剂的组合已被证明在癌症治疗方案中具有重大的进步,但对于难以治疗或对作为单一疗法的常规抗肿瘤剂表现出治疗抗性的癌症的治疗,仍有一些未满足的需求和改善癌症的药物治疗的空间。例如,临床治疗中虽然有以吉西他滨为基础的联合化疗治疗方案,但肿瘤耐药性最终导致胰腺癌预后很差,中位生存期仅为3-6个月,5年生存率小于5%。因此,开发用于递送具有不同作用机制的已知抗癌剂的新型组合途径是本领域的重要进步。虽然涉及具有不同作用机制的抗癌剂组合的方案可以在某些组合的情况下发挥作用,但相同的方式对于抗癌剂的其它组合可能不起作用,且这样的组合可能不总是产生具有有利治疗效果的组合。技术实现要素:技术问题:为了解决现有技术的缺陷,本发明提供了一种抗肿瘤药物组合物。技术方案:本发明提供的一种抗肿瘤药物组合物,包含活性成分和药学上可接受的辅料,所述的活性成分由吉西他滨和a晶型式i所示的酞嗪酮类化合物组成,所述活性成分中吉西他滨和式i所示的酞嗪酮类化合物的质量比为(1-3):5;其中,式i所示的酞嗪酮类化合物的化学名为[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐,化学式为:作为改进,i所示的酞嗪酮类化合物的a晶型,使用cu-ka辐射,其x-射线粉末衍射在衍射角2θ为4.3±0.2°、19.0±0.2°、19.3±0.2°、20.3±0.2°、20.6±0.2°、24.6±0.2°和24.9±0.2°处显示特征峰。本领域知道,用x射线衍射测定化合物的结晶时,由于测定的仪器或测定的条件等的影响,对于所测得的峰会存在一定的测量误差,特别是x-射线衍射图的相对强度随试验条件的变化而变化。例如,2θ值的测定误差可为约±0.2°,相对强度的测定误差可为±20%。因此,在确定每种结晶结构时,应该将此误差考虑在内。可以理解为任何与本申请的a晶型具有基本相同或相似的x-射线衍射图的晶型均属于本申请的范围之内。作为改进,使用cu-ka辐射,其x-射线粉末衍射在衍射角2θ为4.3±0.2°、8.8±0.2°、9.4±0.2°、14.5±0.2°、15.1±0.2°、15.6±0.2°、19.0±0.2°、19.3±0.2°、20.3±0.2°、20.6±0.2°、23.1±0.2°、24.1±0.2°、24.6±0.2°、24.9±0.2°处显示特征峰。作为改进,使用cu-ka辐射,其x-射线粉末衍射在衍射角2θ为4.3±0.2°、8.8±0.2°、9.4±0.2°、14.5±0.2°、15.1±0.2°、15.6±0.2°、19.0±0.2°、19.3±0.2°、20.3±0.2°、20.6±0.2°、23.1±0.2°、24.1±0.2°、24.6±0.2°、24.9±0.2°、30.1±0.2°和30.9±0.2°处显示特征峰。本发明提供的[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型具有如图1所述x-衍射图。作为改进,a晶型式i所示的酞嗪酮类化合物的制备方法,包括以下步骤:步骤a:式(1)的化合物与式(2)的化合物缩合得到式(3)的化合物;步骤b:式(3)的化合物与式(4)的化合物反应得到式(5)的化合物;步骤c:式(5)的化合物脱去保护基得到式(6)的化合物;步骤d:式(6)的化合物与丙烯酰氯反应得到式i化合物;步骤e:式(i)化合物与甲磺酸反应制得式(i)化合物甲磺酸盐,反应路线如下:步骤f:将步骤e所得式(i)化合物甲磺酸盐加入甲醇-丙酮-水混合溶剂中,回流,0-5℃析晶,即得a晶型的[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐。步骤f中所述的甲醇-丙酮-水混合溶剂中甲醇、丙酮和水的体积比为(10~15):2:2。步骤f中所述的甲醇-丙酮-水混合溶剂中甲醇、丙酮和水的体积比为6:1:1。步骤f中所述的甲醇-丙酮-水混合溶剂的使用量为每克[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐使用甲醇-丙酮-水混合溶剂5-15ml。步骤f中所述的甲醇-丙酮-水混合溶剂的使用量为每克[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐使用甲醇-丙酮-水混合溶剂5-10ml。有益效果:与现有技术相比,本发明涉及的药物组合物抗癌疗效好。本发明发现吉西他滨和a晶型式i所示的酞嗪酮类化合物组合后能大大提高抗肿瘤生长的生物活性,二者联用具有抗肿瘤增殖的协同作用,渴望开发成临床上抗胰腺癌的一线用药。附图说明图1是[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的x-射线衍射图。具体实施方式下面对本发明作出进一步说明。实施例1[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯的制备步骤1:称取5-氨基-3,4-二氢异喹啉-2(1h)-甲酸叔丁酯(50mmol)和dipea(100mmol)于反应瓶中,加入二氯甲烷300ml,室温搅拌下缓慢滴加氯甲酸对氯苄酯(51mmol),滴毕,室温下继续搅拌1h,停止反应,浓缩反应混合物,加入乙酸乙酯70ml,稀盐酸水溶液(0.2-0.3n)和饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩得(3,4-二氢异喹啉-2(1h)-甲酸叔丁酯-5-基)-氨基甲酸对氯苄酯,直接用于下一步,esi–ms:[m+h]+m/z417。步骤2:称取3-氟-1-二甲氧基甲基苯(500mmol)于反应瓶中,加入四氢呋喃(800ml)溶解,60℃、氮气保护下,加入s-buli(565mmol),将反应液在-60℃下搅拌1h;称取干冰(50mmol)于另一反应瓶中,加入四氢呋喃(200ml),加入n-buli(5ml),氮气保护下搅拌2h后,加入上述混合物,继续搅拌30min,停止反应,加入水1000ml,用浓盐酸调节ph至2,分离有机相,水相用乙酸乙酯萃取,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,重结晶得到6-氟-2-二甲氧基甲基苯甲酸;称取6-氟-2-二甲氧基甲基苯甲酸(400mmol)、乙酸(93mmol)、肼(600mmol)于反应瓶中,加入异丙醇300ml,氮气保护下,100℃回流反应2h,停止反应,加入乙酸乙酯300ml,水500ml,萃取,合并有机相,无水硫酸钠干燥,悬干,柱层析纯化得8-氟-2h-酞嗪-1-酮,esi–ms:[m+h]+m/z165。步骤3:称取8-氟-2h-酞嗪-1-酮(150mmol)、(3,4-二氢异喹啉-2(1h)-甲酸叔丁酯-5-基)-氨基甲酸对氯苄酯(195mmol)于反应瓶中,加入dmf100ml,55℃下反应过夜,停止反应,加入水100ml,二氯甲烷200ml,萃取,分离有机相,水相继续用二氯甲烷萃取(3*50ml),合并有机相,无水硫酸钠干燥,柱层析纯化得(3,4-二氢异喹啉-2(1h)-甲酸叔丁酯-5-基)-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯。步骤4:称取(3,4-二氢异喹啉-2(1h)-甲酸叔丁酯-5-基)-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯(50mmol)于反应瓶中,加入三氟乙酸20ml,室温下搅拌1h,减压浓缩至干,加入乙酸乙酯80ml,依次用1.5m磷酸氢二钠水溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩得中间体(1,2,3,4-四氢异喹啉-5-基)氨基甲酸-8-氟-2-(2h)-酞嗪-1-酮基苄酯;称取所得中间体(1,2,3,4-四氢异喹啉-5-基)氨基甲酸-8-氟-2-(2h)-酞嗪-1-酮基苄酯(20mmol)于反应瓶中,加入二氯甲烷100ml溶解,0℃下加入diea(40mmol),搅拌30min后,继续在0℃下滴加丙烯酰氯(20mmol),滴毕,室温搅拌3h,停止反应,加水100ml,二氯甲烷萃取(3*50ml),合并有机相,无水硫酸钠干燥,柱层析纯化得[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯。1hnmr(600mhz,cdcl3)(δ,ppm):9.50(s,1h),8.10(s,1h),7.88~7.85(m,1h),7.77~7.75(m,1h),7.54~7.52(m,3h),7.34~7.32(m,2h),7.21~7.18(m,3h),6.57~6.55(m,1h),6.41~6.39(m,1h),4.65(s,2h),4.22(s,2h),3.61~3.59(m,2h),3.13~3.11(m,2h),2.05~2.03(m,3h).esi–ms:[m+h]+m/z513。实施例2:[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐的制备称取实施例1化合物(0.5mmol)于反应瓶中,加丙酮5ml室温下搅拌0.5h,滴加甲磺酸丙酮溶液(含甲磺酸0.55mmol)1ml,滴毕,室温下继续搅拌1h,加压蒸除溶剂,室温下真空干燥获得标题化合物。1hnmr(600mhz,cdcl3)(δ,ppm):9.50(s,1h,重水交换后消失),8.10(s,1h),7.88~7.85(m,1h),7.77~7.75(m,1h),7.54~7.52(m,3h),7.34~7.32(m,2h),7.21~7.18(m,3h),6.57~6.55(m,1h),6.41~6.39(m,1h),5.17(s,2h),4.22(s,2h),3.61~3.59(m,2h),3.13~3.11(m,2h),2.84~2.81(s,3h),2.05~2.03(m,3h).实施例3:[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的制备称取1.0g[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐于反应瓶中,加入12ml体积比为5:1:1的甲醇-丙酮-水混合溶液,回流10min,自然冷却至室温,0-5℃析晶12h,得到[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型。实施例4:[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的制备称取1.0g[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐于反应瓶中,加入10ml体积比为6:1:1的甲醇-丙酮-水混合溶液,回流10min,自然冷却至室温,0-5℃析晶12h,得到[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型。实施例5:[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的制备称取1.0g[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐于反应瓶中,加入7ml体积比为6:1:1的甲醇-丙酮-水混合溶液,回流10min,自然冷却至室温,0-5℃析晶12h,得到[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型。实施例6:[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的制备称取1.0g[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐于反应瓶中,加入5ml体积比为15:2:2的甲醇-丙酮-水混合溶液,回流10min,自然冷却至室温,0-5℃析晶12h,得到[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型。以上实施例3-6制备的[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型,经测量具有基本相同的x射线衍射图谱,见图1。实施例7[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型的稳定性测试称取3份[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型1.0g,分别平铺于培养皿中,1份开口置于光照照度为4500lx±500lx条件下放置10天,1份开口置于室温75%相对湿度(rh)条件下放置10天,1份开口置于室温92.5%相对湿度(rh)条件下放置10天,于第0、5、10天取样,考查最大单杂、总杂质、晶型和外观的变化,实验结果见表1。表1上述实验结果均表明,本发明的[2(1h)-丁-2-烯酰基-3,4-二氢异喹啉-5-基]-氨基甲酸-4-[8-氟-(2h)-酞嗪-1-酮基]苄酯甲磺酸盐a晶型具有良好的化学稳定性和物理稳定性。实验例8体外激酶活性评价制备组合物:组合物1:质量比为1:5的吉西他滨和式i所示的酞嗪酮类化合物;组合物2:质量比为3:5的吉西他滨和式i所示的酞嗪酮类化合物;组合物3:质量比为2:5的吉西他滨和式i所示的酞嗪酮类化合物;对照组1:吉西他滨;对照组2:式i所示的酞嗪酮类化合物。以上组合物和对照组用dmso稀释至10mm后,依次稀释至1um、100nm、10nm、1nm、0.1nm、0.01nm。取每个浓度的化合物溶液10μl至96孔板中,加入90μl1×激酶缓冲液(50mmhepes,ph7.5,0.0015%brij-35,10mmmgcl2,2mmdtt,临用前配制);同时设立dmso对照组和无酶活对照组,均仅含10μldmso和90μl1×激酶缓冲液。各组在室温下混匀10min,然后分别转移5μl至384孔板中;将激酶btk溶于1×激酶缓冲液,配制成2.5×激酶溶液,然后转移10μl2.5×激酶溶液至上述含各浓度化合物的384孔板中;dmso对照组加入10μl2.5×激酶溶液;无酶活对照组加入10μl不含激酶的1×激酶缓冲液。室温下孵育10min;将fam标记的多肽和atp溶于1×激酶缓冲液,配制成2.5×底物溶液,然后转移10μl2.5×底物溶液至上述384孔板中,28℃孵育1hr;各孔中加入25μl(100mmhepes,ph7.5,0.015%brij-35,0.2%coatingreagent#3,50mmedta临用前配制),终止液终止反应;置于labchipezreader上读取转化率数据,并计算抑制率i%,计算公式为i%=(max-conversion)/(max-min)×100,其中max为dmso对照组的转化率,min为无酶活对照组的转化率,conversion为化合物处理组的转化率,数据经xlfit处理,拟合得ic50。ic50值表示与未加化合物处理组相比,化合物抑制50%酶活力时对应的化合物浓度。ic50结果见表1。表1受试化合物ic50(nm)受试化合物ic50(nm)组合物10.8对照组11.0组合物20.8对照组215.1组合物30.6实验例2体外romas细胞活性评价取处于指数生长期状态良好的raji细胞一瓶,收集细胞,低速台式离心机,1500转/min,离心3min。弃上清,用移液器加入5ml完全培养基进行细胞重悬。使用细胞计数仪计数,完全培养基进行稀释,调整细胞密度至5×104个/ml。使用排枪接种于96孔板上,100μl/孔,置恒温co2培养箱中培养24小时。使用纳升加样仪进行化合物加样,72小时后,加cck-8,10μl/孔,2小时后envision酶标仪450nm处检测其吸光值,计算抑制率,并计算ic50,结果见表2.表2受试化合物ic50(nm)受试化合物ic50(nm)组合物11.5对照组12.0组合物21.6对照组216.2组合物31.3当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1