一种应用于宏病毒组高通量测序数据的分析方法与流程
本发明涉及病毒高通量测序领域,具体涉及一种应用于宏病毒组高通量测序数据的分析方法。
背景技术:
可用于宏基因组测序分析的生物信息学工具主要用于鉴定的微生物,但它们通常不考虑病毒。只有一些软件被设计用于分析病毒序列,但是它们在复杂群落中的病毒表征方面效率不高,例如含有细菌,古细菌,真核微生物和病毒的肠道微生物群。在任何情况下,宿主-微生物群相互作用的全面描述不能忽略病毒体内真核病毒的概况,因为病毒对于宿主免疫表型的调节肯定是关键的。
而现有的针对病毒组高通量分析方法存在如下缺陷:
(1)速度慢:现有的工具的运行的所需的时间太长;
(2)没有完整的考虑需求:目前分析流程在两种模式中摇摆,一是基于reads比对数据库,对病毒的种类分类;另一种是基于拼接结果,对病毒进行分类,往往分析的侧重点不同,会使用不同的方法分析;
(3)结果展示单一:分析结果中过于简单,缺少可视化展示的内容。
技术实现要素:
为了克服现有技术所存在的上述缺陷,本发明的目的在于提供一种应用于宏病毒组高通量测序数据的分析方法。
为了实现本发明的目的之一,所采用的技术方案是:
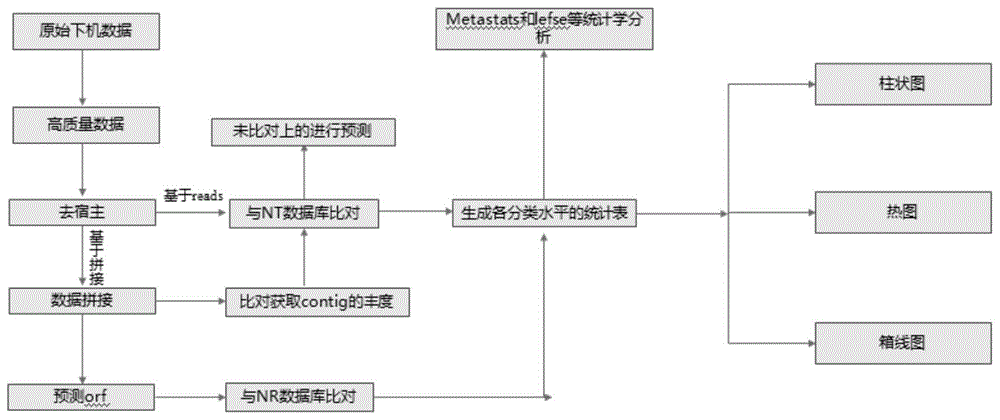
一种应用于宏病毒组高通量测序数据的分析方法,包括如下步骤:
下机数据处理步骤:将下机得到的原始数据,通过cutadapt和trimmomatic软件的去接头,以及质量过滤,得到分析所使用的高质量数据;
高质量数据处理步骤:使用bmtagger软件将上一步得到的高质量数据对其来源,做去宿主,主要;
数据分析步骤:基于拼接分析的流程或基于reads的流程对数据进行分析前的进一步处理;
结果展示步骤:将所述数据分析步骤获得的丰度表或病毒统计结果分别以柱状图,热图,箱线图做可视化展示;
差异分析步骤:将所述丰度表或病毒统计结果以metastats和lefse的方法做组与组之间的差异分析,获取差异的病毒和/或基因;
其中,所述数据分析步骤中,基于拼接分析的流程为:
使用拼接软件spades对去掉宿主后剩余的数据进行拼接,得到拼接结果contig和scaffold;
使用软件bowtie2将用于拼接的reads比对到contig上,并使用软件soap.coverage计算contig的丰度;
使用软件metagenemark对拼接得到的contig预测orf,得到预测的基因;
使用blast软件将上述拼接好的contig与ncbi-nt数据库进行比对,获取对应的病毒注释结果;
使用diamond软件将上述预测好的基因与ncbi-nr数据库进行比对,获取到对应的基因的注释结果;
将病毒注释的结果以及基因的注释结果与对应的contig丰度关系,制作对应的丰度表;
所述数据分析步骤中,基于reads的流程为:
使用blast软件将上述过滤好的reads与ncbi-nt数据库进行比对,获取对应的病毒注释结果;
使用virfinder软件对未注释的reads进行预测是否为未知的病毒;
统计注释上的病毒的数量,将统计结果制作成统计表。
在本发明的一个优选实施例中,所述的一种应用于宏病毒组高通量测序数据的分析方法,更具体地,包括如下步骤:
第一步:将下机得到的原始数据,通过cutadapt和trimmomatic软件的去接头,以及质量过滤,得到分析所使用的高质量数据;
第二步:使用bmtagger软件将上一步得到的高质量数据对其来源,做去宿主,主要;
第三步:使用拼接软件spades对去掉宿主后剩余的数据进行拼接,得到拼接结果contig和scaffold;
第四步:使用软件bowtie2将用于拼接的reads比对到contig上,并使用软件soap.coverage计算contig的丰度;
第五步:使用软件metagenemark对拼接得到的contig预测orf,得到预测的基因;
第六步:使用blast软件将上述拼接好的contig与ncbi-nt数据库进行比对,获取对应的病毒注释结果;
第七步:使用diamond软件将上述预测好的基因与ncbi-nr数据库进行比对,获取到对应的基因的注释结果;
第八步:将病毒注释的结果以及基因的注释结果与对应的contig丰度关系,制作对应的丰度表;
第九步:将对应的丰度表分别以柱状图,热图,箱线图做可视化展示;
第十步:将对应的丰度表以metastats和lefse的方法做组与组之间的差异分析,获取差异的病毒或者基因。
在本发明的一个优选实施例中,所述的一种应用于宏病毒组高通量测序数据的分析方法,更具体地,包括如下步骤:
第一步:将下机得到的原始数据,通过cutadapt和trimmomatic软件的去接头,以及质量过滤,得到分析所使用的高质量数据;
第二步:使用bmtagger软件将上一步得到的高质量数据对其来源,做去宿主,主要;
第三步:使用blast软件将上述过滤好的reads与ncbi-nt数据库进行比对,获取对应的病毒注释结果;
第四步:使用virfinder软件对未注释的reads进行预测是否为未知的病毒;
第五步:统计注释上的病毒的数量,将统计结果制作成统计表;
第六步:将对应的病毒统计结果分别以柱状图,热图,箱线图做可视化展示;
第七步:将对应的病毒统计结果以metastats和lefse的方法做组与组之间的差异分析,获取差异的病毒。
本发明的主要创新点在于:
分析内容全面:同时考虑到了两个常见的分析方法,一种基于reads分析,一种基于拼接结果进行分析,能够让适应各种分析的需求;
运行速度提升:本分析方法能够比现有的分析方法及软件快2到3倍;
结果信息全面:本分析结果中,除了病毒的注释信息外,还将给出相对应的可视化结果,便于查看结果。
附图说明
图1为本发明的流程示意图。
图2为现有技术数据图表示意图。
图3为本发明数据图表示意图1。
图4为本发明数据图表示意图2。
图5为本发明数据图表示意图3。
图6为本发明metastats的结果示意图。
图7为本发明lefse的结果示意图。
图8为本发明lefse柱状图示意图。
具体实施方式
以下通过实施例对本发明作进一步的说明,但这些实施例不得用于解释对本发明的限制。
实施例1:基于拼接分析的流程
1.将原始下机得到的数据,运行软件包cutadapt,去除“agatcggaag”接头,比对错配率小于10%,比对上的长度必须大于10bp,最多去除3次,去除后reads的长度不短于50bp,然后使用质量过滤trimmomatic对去除接头的后数据进行质量过滤,参数为以滑动窗口法过滤,窗口大小10bp,步长5bp,窗口的平均质量不低于q20,且最后过滤完成的read长度不低于50bp。
2.利用去宿主软件bmtagger(采用默认参数)将上一步获得的序列快速映射到宿主核酸数据库(如果是人的样本,则为人的核酸数据库;如果是动物的样本,则为动物的核酸数据库)上,将不能映射的序列保存作后续分析。
17.在核酸水平利用宏基因组学拼接软件spades(默认参数基础上增加—meta参数)对由上一步获得的不能映射到宿主核酸序列上的序列进行拼接。
3.使用基因预测软件metagenemark(默认参数)对拼接得到的contig预测orf,得到预测的基因;
4.将上述步骤获得的拼接序列与准备好的ncbi-nt进行比对,由ncbi的blast软件包进行,采用参数“-evalue1e-5-outfmt6-num_alignments100”。
5.使用diamond软件将上述预测好的基因与ncbi-nr数据库进行比对,获取到对应的基因的注释结果,采用参数“-top10-f6-e1e-5”。
21.利用blast2lca(默认参数加上-levels="superkingdom:norank:order:family:subfamily:genus:species")将blast的结果转换成物种组成信息,采用的最近共同祖先算法(lca,latestcommonancestry)映射到分类单元.
6.使用序列比对软件bowtie2(默认参数)将用于拼接的reads比对到contig上,并使用软件soap.coverage(默认参数)计算contig的丰度。
7.将上述步骤得到的contig的丰度,将之与其对应的物种注释的结果以及蛋白的注释结果,使用awk软件制作成丰度表格,用于后面的可视化展示,以及差异分析。
8.将对应的丰度表使用r软件的ggplot2的软件包,将上述得到的统计结果,在物种方面,以对应的目,科,属,种层次绘制柱状图,热图,箱线图,将功能层次截取丰度前30的基因注释结果绘制柱状图,热图,箱线图。
9.将上述得到的统计结果,以对应的目,科,属,种层次以及所有的功能注释结果以metastats和lefse的方法做组与组之间的差异分析,获取差异的病毒或功能;metastats的筛选条件为pvalue要小于0.05且qvalue要小于0.01,而lefse的筛选条件为wilcoxon秩和检验的pvalue小于0.05,且之后的lda线性判别的ldascore要大于2。
实施例2-基于reads的流程
1.将原始下机得到的数据,运行软件包cutadapt,去除“agatcggaag”接头,比对错配率小于10%,比对上的长度必须大于10bp,最多去除3次,去除后reads的长度不短于50bp,然后使用质量过滤trimmomatic对去除接头的后数据进行质量过滤,参数为以滑动窗口法过滤,窗口大小10bp,步长5bp,窗口的平均质量不低于q20,且最后过滤完成的read长度不低于50bp。
2.利用去宿主软件bmtagger(采用默认参数)将上一步获得的序列快速映射到宿主核酸数据库(如果是人的样本,则为人的核酸数据库;如果是动物的样本,则为动物的核酸数据库)上,将不能映射的序列保存作后续分析。
3.将上述步骤获得的上述得到核酸序列与准备好的ncbi-nt进行比对,由ncbi的blast软件包进行,采用参数“-evalue1e-5-outfmt6-num_alignments100”。
4.利用blast2lca(默认参数加上-levels="superkingdom:norank:order:family:subfamily:genus:species")将blast的结果转换成物种组成信息,采用的最近共同祖先算法(lca,latestcommonancestry)映射到分类单元。
5.使用virfinder软件(默认参数)对未注释的reads进行预测是否为未知的病毒;
6.将上述步骤得到注释结果,统计注释到同一种物种的丰度,将之与其对应的物种注释的结果,使用awk软件制作成丰度表格,并将丰度矫正成rpm(readspermillion)用于后面的可视化展示,以及差异分析。
7.将对应的丰度表使用r软件的ggplot2的软件包,将上述得到的统计结果,在物种方面,以对应的目,科,属,种层次绘制柱状图,热图,箱线图。
8.将上述得到的统计结果,以对应的目,科,属,种层次以metastats和lefse的方法做组与组之间的差异分析,获取差异的病毒;metastats的筛选条件为pvalue要小于0.05且qvalue要小于0.01,而lefse的筛选条件为wilcoxon秩和检验的pvalue小于0.05,且之后的lda线性判别的ldascore要大于2。
而现有技术当中的数据图表很单一,比如:
像virusseeker这种宏病毒组分析软件,最后的统计结果,只是给一个病毒统计的统计表。如图2所示。缺乏一些可视化的结果,本分析方法主要在于在得到类似的病毒统计注释表格后,还增加了一些可视化的图片,如图3-5所示。
此外除了图表外,增加metastats和lefse差异的差异分析,如图6、7、8所示。
而在运行的速度方面:virusseeker依旧使用一些老旧perl程序,这会导致遭遇大量的数据时,运行周期较长,速度较慢。
且本发明与现有技术的区别在于:
virusseeker使用prinseq用于做质量过滤,而本软件使用trimmomatic,在保证处理数据的准确性的前提下,此软件的运行效率比prinseq会高很多。
virusseeker使用是的perl程序解析blast的结果,而本软件则是使用的blast2lca解析,效率比之高了很多。
- 还没有人留言评论。精彩留言会获得点赞!