抗体PROTAC偶联物的制作方法
本发明是关于基于adc及protac技术的新颖治疗剂。
背景技术:
抗体长期以来一直是基础研究以及医学用途中不可或缺的工具,因为其对靶抗原具有高度特异性及亲和力。抗体的一个关键特征是其高特异性及其结合靶抗原的能力,对其进行标记以便利用补体依赖性细胞毒性(cdc)或抗体依赖性细胞介导的细胞毒性(adcc)去除。抗体也可通过结合靶抗原且抑制靶抗原的功能而赋予治疗益处。然而,许多针对肿瘤特异性抗原的未修饰抗体通常缺乏治疗活性。尽管一些抗体可以替代地成功地用作导向导弹从而传递以抗体药物偶联物(adc)形式的有效细胞毒性药物,但许多adc具有有限的治疗潜力并且可能需要进一步改良。
蛋白水解靶向嵌合体(proteolysistargetingchimera,protac)是一种双头分子,能够通过诱导选择性细胞内蛋白水解来除去不需要的蛋白质。protac由两个蛋白质结合部分组成,一个用于结合e3泛素连接酶,而另一个用于结合靶蛋白。通过结合两种蛋白质,protac将靶蛋白带至e3连接酶,导致靶蛋白的标记(即泛素化),随后被蛋白酶体降解。
泛素化涉及三个主要步骤:活化、偶联及连接,分别由泛素活化酶(e1)、泛素偶联酶(e2)及泛素连接酶(e3)进行。此连续级联的结果是将泛素与靶蛋白共价结合。泛素化的蛋白质最终会由蛋白酶体降解。
protac技术于2001年首次被描述(sakamoto等人,“protacs:chimericmoleculesthattargetproteinstotheskp1-cullin-fboxcomplexforubiquitinationanddegradation”,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica.98(15):8554-9)。自此,该技术已被用于几种药物设计:pvhl、mdm2、β-trcp1、小脑蛋白(cereblon)、及c-iap1。虽然这些现有技术的protac药物非常有用,但仍需要更好的protac药物。
技术实现要素:
本发明的实施方式是关于支链抗体-protac偶联物(apc)。本发明的支链抗体-protac偶联物结合了adc及protac两种方法的优点,并且与直链形式的apc相比,支链形式的apc在开发或加工中具有若干益处。在本发明的支链抗体-protac偶联物中,常规adc中的负载(药物)经protac置换并连接至protac分子的连接子部分。这些新治疗剂的选择性高,毒性低,使用更安全,且体内半衰期更长。
本发明的一个方面是关于免疫偶联物。根据本发明的一个实施方式的免疫偶联物具有式(i):ab-[l2-(a-l1-b)m]n,其中:(a)ab是抗体或其结合片段;(b)l1及l2各自独立地是连接子,其中,l1及l2可以相同或不同;(c)a是靶蛋白配体/结合物;(d)b是泛素连接酶配体/结合物,且(e)n及m各自独立地是1至8的整数。
根据本发明的一些实施方式,靶蛋白可以是激酶、g蛋白偶联受体、转录因子、磷酸酶、及ras超家族成员。
通过以下描述及所包括的附图,本发明的其他方面将变得显而易见。apc及支链的结构
附图说明
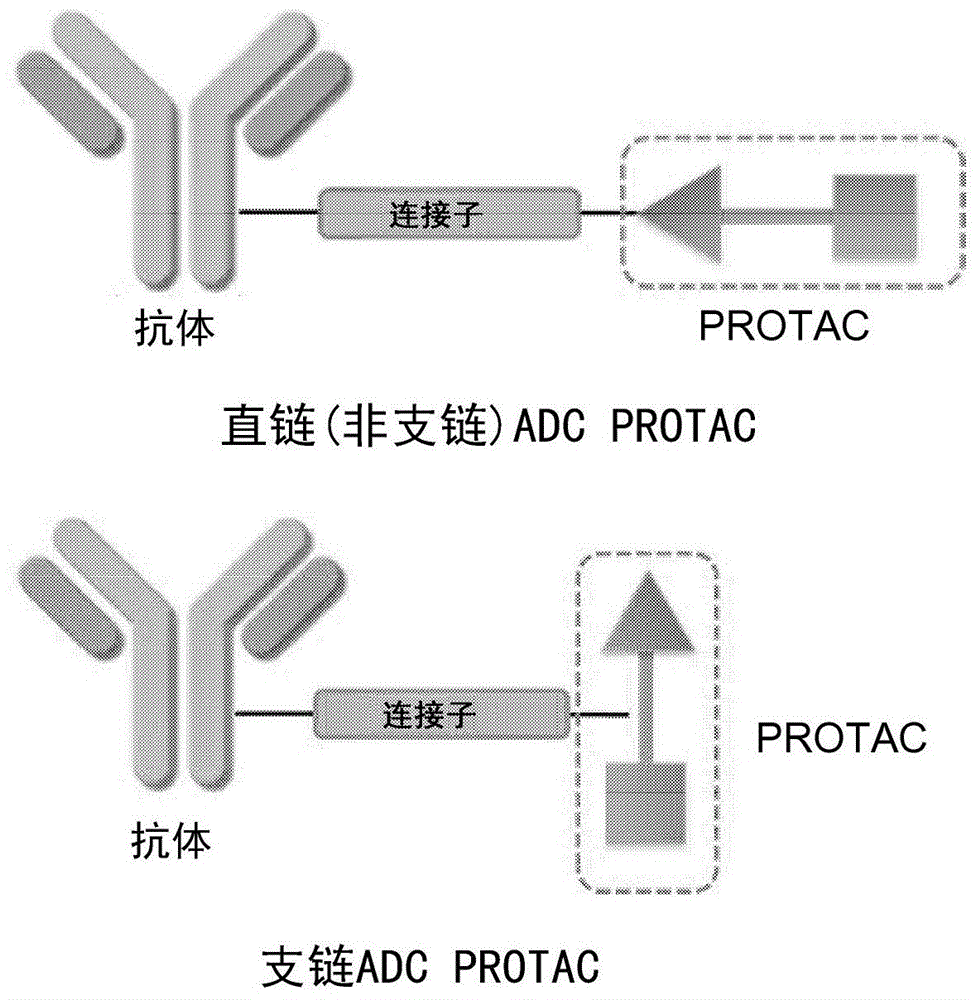
图1显示说明直链(非支链)apc的结构及支链apc的结构的示意图。
图2显示如用sds-page电泳及western印迹所分析的,在bt-474乳腺癌细胞培养物中,各种浓度的arv-825及本发明化合物5(但并非本发明化合物7)对brd4降解的促进作用。arv-825是已知的小分子的靶向brd4的protac。本发明化合物5是arv-825的支链形式。结果显示化合物5具有与arv-825对brd4相当的降解活性。相反,本发明化合物7,其是具有溶酶体可切除的另外的二肽(缬氨酸-瓜氨酸)的化合物5,在高达1μm的化合物7处理时不能降解brd4蛋白。此结果可能源于以下事实:由于缬氨酸-瓜氨酸二肽而具有相对较低渗透性的化合物7不能有效地内化至细胞中。brd4及akt标记brd4及akt条带的位置,且肌动蛋白用作加载对照。
图3显示实施例1的在her2阳性bt-474乳腺癌细胞而非her2阴性mda-mb-231乳腺癌细胞中表达出特异性brd4蛋白质降解活性。实施例1是支链曲妥珠单抗-化合物7免疫偶联物(即,具有曲妥珠单抗作为抗体及化合物7作为protac的支链apc)。此外,这种支链apc不会引起akt蛋白或肌动蛋白的降解。
图4显示通过经arv-825中的a结合物连接的直链apc的合成方案。
具体实施方式
本发明的实施方式涉及支链apc。支链apc经由protac的连接子部分将抗体与protac偶联。本发明的支链apc可以被视为抗体-药物偶联物(adc)的类似物,其中,常规adc中的负载(药物)经protac置换,protac经由protac中的连接子与抗体偶联。即,在本发明的支链apc中,抗体(或其结合片段)经由连接子(l2)与protac经由protac的连接子部分(l1)共价连接,而非经由靶蛋白结合部分(a)或泛素连接酶结合物(b)连接。抗体上的共价连接可以在蛋白质部分(例如,恒定区或可变区)上或在碳水化合物(糖部分)上。
图1显示说明直链apc与支链apc之间的差异的示意图。支链apc优于直链apc的优点可能包括以下各项:
1.维持protac部分中的靶配体(a)及e3连接酶配体(b)的原始结构,使得a及b的结合亲和力不会改变,因为抗体连接于protac部分中的连接子(l1),而非连接在protac部分的任一端(a或b)。
2.连接基团在连接子上的结构修饰比在配体(a或b)上的修饰容易得多,并且两个连接子(l1及l2)之间的连接更灵活。
3.连接子的修饰适用于大多数apc。因此,可以设计连接子以使用共同的偶联官能团,使得不同的抗体部分可以容易地与相同的protac偶联,或者相同的抗体可以容易地与不同的protac偶联。
根据本发明实施方式的支链apc可以用下式(i)表示:
其中:
(a)ab是抗体或其结合片段;
(b)l1及l2独立地是连接子,其中,l1是protac部分内的连接子(即protac=a-l1-b),且l2是经由protac的连接子部分(l1)连接抗体与protac的连接子;
(c)a是靶配体/结合物(即,靶蛋白的结合物,该靶蛋白可以是激酶、g蛋白偶联受体、转录因子、磷酸酶、ras超家族成员等);
(d)b是泛素连接酶配体/结合物,其中,泛素连接酶可以是e2或e3泛素连接酶,且
(e)n及m独立地是1至8的整数。
本发明的支链apc具有adc及protac两者的优点,并代表一类新的治疗剂。术语“支链”是指上式(i)中所示的结构,其中,抗体-l2连接子与protac中的l1连接子偶联。其中抗体-l2连接子连接至protac的任一末端(a或b)的其他类型的apc将称为“非支链的”或“直链的”。这些新的支链apc的选择性高,体内半衰期长,治疗窗口大,适用性广,且使用更安全。此外,这些支链apc比非支链(直链)apc更容易合成。
抗体-药物偶联物(adc)是一类治疗剂,其中,药物(或负载)与抗体或其抗原结合片段连接。adc中的抗体与选定的靶(通常是细胞上的靶)结合,借此将药物带至靶附近,从而产生高选择性的治疗效果。adc的实例可以是靶向癌细胞上表达的蛋白质的抗体,并且负载可以是细胞毒性剂(例如紫杉醇)。
由于存在抗体,adc是大分子,分子量通常为约150kda或更高。因此,肾脏过滤不会消除adc。此外,抗体恒定区包括与肾中受体相互作用的位点,其可以将抗体运送且再循环回至循环中。因此,抗体的体内半衰期长,通常为数周。此外,抗体可以容易地被细胞内化,使得负载极有效地传递至细胞中。由于adc具有特异性及长效性,因此是有前景的治疗剂。然而,adc的作用依赖于负载,其并不像催化剂一样起作用。因此,需要足够量的负载来杀死或抑制靶蛋白或细胞。过载可能会导致毒性。
protac由两个蛋白质结合部分组成,一个用于结合e3泛素连接酶,而另一个用于结合靶蛋白。protac可以结合其靶蛋白并将其带至e3泛素连接酶。在三级复合物形成后,e3泛素连接酶将泛素转移至靶蛋白的表面赖氨酸,产生泛素化的靶蛋白,其注定要由蛋白酶体机器降解。泛素化后,protac经释放并继续寻找用于泛素化及降解的靶蛋白。因此,protac像催化剂一样起作用,且少量的protac可以取得实质性的结果。
在上式(i)中,a组分是与欲降解的靶蛋白结合的基团。a组分可包括特异性结合于靶蛋白的任何部分。以下是小分子靶蛋白结合部分的非限制性实例:hsp90抑制剂、激酶抑制剂、mdm2抑制剂、靶向含人类bet溴结构域(bromodomain)的蛋白质的化合物、hdac抑制剂、人类赖氨酸甲基转移酶抑制剂、血管生成抑制剂、免疫抑制化合物及靶向芳基烃受体(ahr)的化合物等。下面描述的组合物举例说明这些类型的小分子靶蛋白结合部分的一些成员。这种小分子靶蛋白结合部分也包括这些组合物的药学上可接受的盐、对映异构体、溶剂合物及多晶型物,以及可以靶向所关注蛋白质的其他小分子。
通常,靶蛋白可包括例如结构蛋白、受体、酶、细胞表面蛋白、与细胞功能相关的蛋白质等。因此,adc-protac的a组分可以是结合蛋白质靶的任何肽或小分子,如foxol、hdac、dp-1、e2f、abl、ampk、brk、brski、brsk2、btk、camkk1、camkkα、camkkβ、rb、suv39hi、scf、p19ink4d、gsk-3、pi8ink4、myc、细胞周期蛋白e、cdk2、cdk9、cdg4/6、环蛋白d、pl6ink4a、cdc25a、bmi1、scf、akt、chk1/2、c1δ、ck1γ、c2、clk2、csk、ddr2、dyrk1a/2/3、ef2k、eph-a2/a4/b1/b2/b3/b4、eif2a3、smad2、smad3、smad4、smad7、p53、p21cipl、pax、fyn、cas、c3g、sos、tal、raptor、rack-1、crk、rapl、rac、kras、nras、hras、grb2、fak、pi3k、spred、spry、mtor、mpk、lkbl、pak1/2/4/5/6、pdgfra、pyk2、src、srpk1、plc、pkc、pka、pkbα/β、pkcα/γ/ζ、pkd、plkl、prak、prk2、wave-2、tsc2、dapki、bad、imp、c-tak1、takl、taol、tbk1、tesk1、tgfbr1、tie2、tlk1、trka、tssk1、ttbk1/2、ttk、tpl2/cotl、mek1、mek2、pldlerk1、erk2、erk5、erk8、p90rsk、pea-15、srf、p27kip1、tifla、hmgn1、er81、mkp-3、c-fos、fgf-r1、gck、gsk3β、her4、hipk1/2/3/、igf-1r、cdc25、ubf、lamtor2、statl、stao、creb、jak、src、pten、nf-κb、hecth9、bax、hsp70、hsp90、apaf-1、cytoc、bcl-2、bcl-xl、smac、xiap、半胱天冬酶-9、半胱天冬酶-3、半胱天冬酶-6、半胱天冬酶-7、cdc37、tab、ikk、tradd、traf2、r1p1、flip、takl、jnk1/2/3、lck、a-raf、b-raf、c-raf、mos、mlk1/3、mn1/2、msk1、mst2/3/4、mpsk1、mekki、mek4、mel、ask1、mink1、mkk1/2/3/4/6/7、ne2a/6/7、nuak1、osr1、sap、stk33、syk、lyn、pdk1、phk、pim1/2/3、紫杉醇-1、mtorcl、mdm2、p21wafl、细胞周期蛋白dl、lamlna、tpl2、myc、连环素、wnt、ikk-β、ikk-γ、ikk-α、ikk-ε、elk、p65rela、iraki、ira2、irak4、irr、fadd、traf6、traf3、mkk3、mkk6、rock2、rsk1/2、sgk1、smmlck、sik2/3、ulk1/2、vegfr1、wnkl、yes1、zap70、map4k3、map4k5、mapklb、mapkap-k2k3、p38α/β/δ/γmapk、极光(aurora)a、极光b、极光c、mcak、clip、mapkapk、fak、mark1/2/3/4、mucl、shc、cxcr4、gap-1、myc、β-连环素/tcf、cbl、brm、mcl-1、brd2、brd3、brd4、ar、ras、erbb3、egfr、ire1、hpk1、ripk2、及erct,包括列出的这些靶蛋白的所有变体、突变、剪接变体、插入缺失及融合物。
b组分是结合e3泛素连接酶的基团。e3泛素连接酶(其中超过600种在人类中已知)赋予受体对泛素化的特异性。存在结合这些连接酶的已知配体。如本文所述,e3泛素连接酶结合基团可以是可以结合e3泛素连接酶的肽或小分子。e3泛素连接酶的实例包括:vonhippel-lindau(vhl);小脑蛋白、xiap、e3a;mdm2;细胞分裂后期促进复合物;ubr5(eddi);socs/bc-box/elobc/cul5/ring;lnxp80;cbx4;cbll1;hace1;hectd1;hectd2;hectd3;hecw1;hecw2;herc1;herc2;herc3;herc4;huwe1;itch;nedd4;nedd4l;ppil2;prpf19;pias1;pias2;pias3;pias4;ranbp2;rnf4;rbx1;smurf1;smurf2;stub1;topors;trip12;ube3a;ube3b;ube3c;ube4a;ube4b;uboxs;ubr5;wwp1;wwp2;parkin;a20/tnfaip3;amfr/gp78;ara54;β-trcpl/btrc;brca1;cbl;chip/stub1;e6;e6ap/ube3a;f-box蛋白15/fbxo15;fbxw7/cdc4;grail/rnf128;hoip/rnf31;ciap-1/hiap-2;ciap-2/hiap-1;ciap(pan);itch/aip4;kap1;march8、mindbomb1/mib1;mindbomb2/mib2;murf1/trim63;ndfip1;nedd4;nlel;parkin;rnf2;rnf4;rnf8;rnf168;rnf43;sart1;skp2;smurf2;traf-1;traf-2;traf-3;traf-4;traf-5;traf-6;trims;trim21;trim32;ubr5;及znrf3。
示例性e3泛素连接酶是vonhippel-lindau(vhl)肿瘤抑制因子,其是e3连接酶复合物vcb-cul2的底物识别亚基。vccb-cul2复合物由vhl、延伸蛋白(elongin)b及c、cul2及rbx1组成。vhl的主要底物是缺氧诱导因子let(hif-let),其是转录因子,响应于低氧水平而上调基因,诸如促血管生成生长因子vegf及诱生红血球的细胞激素红血球生成素。结合vhl的化合物可以是羟脯氨酸化合物,诸如wo2013/106643中揭示的那些,以及us2016/0045607、wo2014187777、us20140356322、及us9,249,153中描述的其他化合物。
另一种示例性e3泛素连接酶是小脑蛋白。小脑蛋白(cereblon)是一种蛋白质,其与受损的dna结合蛋白1(ddb1)、cullin-4a(cul4a)及cullin1调节剂(roc1)形成e3泛素连接酶复合物。该复合物使许多其他蛋白质泛素化。靶蛋白的小脑蛋白泛素化导致纤维母细胞生长因子8(fgf8)及纤维母细胞生长因子10(fgf10)的水平增加。fgf8又调节许多发育过程,例如肢体及听囊形成。在没有小脑蛋白的情况下,ddb1与ddb2形成复合物,其作为dna损伤结合蛋白起作用。已知沙利度胺(thalidomide)、来那度胺(lenalidomide)、泊马度胺(pomalidomide)及其类似物与小脑蛋白结合。结合小脑蛋白的其他小分子化合物也是已知的,例如us2016/0058872及us2015/0291562中公开的化合物。此外,邻苯二甲酰亚胺与结合物(例如bet溴结构域的拮抗剂)的偶联可以为protac提供高选择性的小脑蛋白-依赖的bet蛋白质降解。winter等人,science,2015年6月19日,第1376页。这样的protac可以与本文所述的抗体偶联以形成apc。
protac的特异性依赖于其结合靶蛋白进行降解的靶配体。若特异性不高,则此可能导致脱靶效应(副作用)。此外,protac通常是小分子(mw约1000)。其依赖于扩散进入细胞,效率较低(特别在分子量约为1000的情况下)。此外,因为其是小分子,由于肾脏过滤通常体内半衰期较短。因此,可能需要以更高的给药频率给予protac。
本发明的抗体-protac偶联物(apc)在大小上与抗体或adc相似,其体内半衰期较长。因此,本发明的apc也具有较长的体内半衰期(例如,数周),并且由于抗体的存在可以通过内化进入细胞。此外,apc具有双重选择性:一种来自抗体,而另一种来自protac中的靶蛋白结合物。例如,本发明的apc中的抗体可以结合癌细胞上的特定抗原,接着apc通过内化进入细胞。一旦进入细胞,protac部分中的靶蛋白结合物就会找到靶蛋白并将其带至e3泛素连接酶中进行泛素化。泛素化的靶蛋白经过标记以便由蛋白酶体降解。因此,本发明的apc的选择性高且副作用少。
此外,本发明的apc具有催化作用模式的优点,类似于protac。因此,apc的治疗有效剂量可以更低,并且由于其更长的体内半衰期,其可以以更低的频率给予。这些性质使得本发明的apc更具特异性且使用更安全。
下表1总结并比较adc、protac及apc的一些特征。
表1
因此,apc形式是新颖的并且代表有希望的新治疗剂方法。此方法通常可应用于与任何失调相关的任何靶蛋白。(参见crews等人,「proteolysis-targetingchimeras:inducedproteindegradationasatherapeuticstrategy」,acschem.biol.2017,12(4),892-898)。
本发明的实施方式可以应用于引起疾病或失调的任何靶蛋白,通过获得抗体且接着使用该抗体与protac偶联(如与e3泛素连接酶配体或抑制剂偶联的靶蛋白结合物)。抗体针对在含有靶蛋白的细胞上表达的抗原。protac中的靶蛋白结合物将视所靶向的蛋白质而定。例如,对于靶酶(例如激酶),可以设计抑制剂作为配体/结合物。
对于e3泛素连接酶配体/结合物,已知几种分子结合各种e3泛素连接酶。实例包括:
nutlin衍生物结合mdm2(双微2同系物;也称为e3泛素-蛋白质连接酶mdm2),其是p53肿瘤抑制因子的负调节剂。mdm2具有作为识别p53肿瘤抑制因子的n端反式活化结构域(tad)的e3泛素连接酶及作为p53转录活化的抑制剂的功能。贝他定(bestatin)(乌苯美司(ubenimex))接合ciap1(细胞凋亡蛋白-1的抑制剂)。imid沙利度胺及其衍生物泊马度胺(pomalidomide)及来那度胺(lenalidomide)结合小脑蛋白。
以下描述使用特定实施例来说明本发明的实施方式。这些实施例使用溴结构域及额外末端结构域(bet)蛋白家族作为靶蛋白,并使用曲妥珠单抗(trastuzumab)作为抗体。然而,在这些实施例中使用的任何特定的bet抑制剂、泛素连接酶3抑制剂及曲妥珠单抗仅用于说明,且不应解释为限制本发明的范围。本领域技术人员将理解,在不脱离本发明的范围的情况下,其他修改及变化是可能的。
溴结构域及额外末端结构域(bet)蛋白家族,包括brd2、brd3及brd4,通过控制组蛋白乙酰化依赖性染色质复合物的组装而在许多细胞过程中发挥关键作用,包括发炎基因表达、有丝分裂及病毒/宿主相互作用。bet蛋白的抑制剂可逆地结合溴结构域或bet蛋白:brd2、brd3、brd4、及brdt。其可以阻止bet蛋白与乙酰化组蛋白及转录因子之间的蛋白质-蛋白质相互作用。因此,bet抑制剂具有抗癌、免疫抑制及其他作用。在apc中使用bet抑制剂将靶向bet家族蛋白质进行泛素化,借此引起蛋白酶体消除bet家族蛋白质。
以下描述本发明的一些实施方式的细节。然而,这些细节仅用于说明,并且本领域技术人员将理解,在不脱离本发明的范围的情况下,其他修改及变化是可能的。
实施例1:曲妥珠单抗-brd4-protac-1的制备
化合物2的合成
在此实施例中,bet抑制剂是otx015,其是brd2、brd3及brd4的口服生物可利用的小分子抑制剂(ec50=10至19nm)。otx015下调c-myc表达并诱导细胞周期停滞及细胞凋亡。因此,其对多种实体肿瘤及白血病具有抗增殖作用。
化合物2:向otx015(1)(0.2mmol)及1-溴-2-(2-溴乙氧基)乙烷(1mmol)在二甲基甲酰胺(5ml)中的混合物中添加碳酸钾(0.6mmol)。将混合物在50℃下搅拌24小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。通过柱层析用甲醇:二氯甲烷(1:19)纯化残余物,得到黄色固体化合物2(58%产率)。1hnmr(600mhz,氯仿-d):δ7.45(d,j=9.1hz,2h),7.38(d,j=8.6hz,2h),7.28(d,j=8.6hz,2h),6.78(d,j=9.1hz,2h),4.71(dd,j=8.0,6.2hz,1h),4.06(dd,j=5.4,4.5hz,2h),3.86-3.82(m,4h),3.78(dd,j=14.5,8.0hz,1h),3.57(dd,j=14.5,6.2hz,1h),3.47(t,j=6.2hz,2h),2.66(s,3h),2.38(d,j=0.6hz,3h),1.65(d,j=0.6hz,3h)。lcms(esi):m/z[c33h37cln6o4s+h]+的计算值642.08,实验值642.52[m+h]+。
化合物4的合成
化合物4:向泊马度胺(3)(0.2mmol)及(2-(2-胺基乙氧基)乙基)胺基甲酸第三丁酯(0.22mmol)在dmf(5ml)中的溶液中添加n,n-二异丙基乙胺(0.4mmol)。将反应混合物在90℃下搅拌12小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。在闪蒸塔上用乙酸乙酯:己烷(2:3)纯化后,将黄色残余物溶于二氯甲烷中,且添加三氟乙酸(1ml)。将混合物在室温下搅拌0.5小时。之后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。残余物通过柱层析用甲醇:二氯甲烷(1:9)纯化,得到黄色固体化合物4。1hnmr(600mhz,氯仿-d)δ7.35(t,j=7.8hz,1h),6..93(d,j=7.0hz,1h),6.78(d,j=8.6hz,1h),6.38(d,j=5.1hz,1h),4.81(dd,j=11.6,5.1hz,1h),3.62(d,j=4.0hz,2h),3.56(d,j=4.5hz,2h),3.33(d,j=4.1hz,2h),3.05-3.04(m,2h),2.71-2.53(m,3h),2.01-1.93(m,1h)。lcms(esi):m/z[c33h37cln6o4s+h]+的计算值361.14,实验值361.22[m+h]+。
化合物5的合成
化合物5:向化合物2(0.2mmol)、化合物4(0.6mmol)及碘化钾(0.2mmol)在乙腈/二甲基甲酰胺(3:1,4ml)中的溶液中添加碳酸钾(0.6mmol)。将反应混合物在50℃下搅拌72小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。通过柱层析用甲醇:二氯甲烷(1:12)纯化残余物,得到黄色固体化合物5(19.7%产率)。1hnmr(600mhz,氯仿-d)δ7.53-7.36(m,5h),7.36-7.29(m,2h),7.09(dd,j=7.0,3.7hz,1h),6.88(d,j=8.7hz,1h),6.82(dd,j=8.7,1.4hz,2h),4.89(dt,j=12.5,6.1hz,1h),4.66(ddd,j=7.9,6.0,3.5hz,1h),4.10-4.02(m,2h),3.84-3.58(m,9h),3.55-3.41(m,2h),3.41-3.30(m,2h),3.02-2.91(m,3h),2.87-2.68(m,3h),2.67(s,3h),2.40(s,3h),2.10-2.07(m,1h),1.67(s,3h)。lcms(esi):m/z[c33h37cln6o4s+h]+的计算值922.30,实验值922.49[m+h]+。
化合物7的合成
化合物7:向化合物5(0.2mmol)及mal-c5-vc-pab-pnp化合物6(0.2mmol)在二甲基甲酰胺(5ml)中的溶液中添加羟基苯并三唑(0.4mmol)及吡啶(0.4mmol)。将反应混合物在室温下搅拌72小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。通过柱层析用甲醇:二氯甲烷(1:16)纯化残余物,得到黄色固体化合物7(46.3%产率)。1hnmr(600mhz,氯仿-d)δ7.55-7.40(m,7h),7.33(d,j=7.2hz,2h),7.25-7.19(m,2h),7.08(d,j=7.0hz,1h),6.92-6.83(m,1h),6.79-6.68(m,2h),6.68-6.65(m,2h),5.07-5.00(m,2h),4.77-4.71(m,1h),4.71-4.60(m,1h),4.37-4.29(m,1h),4.04-3.96(m,1h),3.93-3.85(m,1h),3.77-3.34(m,21h),3.28-3.14(m,1h),3.11-3.02(m,1h),2.93-2.71(m,3h),2.69(s,3h),2.42(s,3h),2.04-2.01(m,1h),1.91-1.81(m,2h),1.77-1.70(m,1h),1.69(s,3h),1.62-1.54(m,4h),1.33-1.25(m,5h),0.91-0.87(m,6h)。lcms(esi):m/z[c33h37cln6o4s+h]+的计算值1520.58,实验值1521.14[m+h]+。
曲妥珠单抗-brd4-protac1的偶联
向曲妥珠单抗1mg(5.0mg/ml)在缓冲液(25mm硼酸钠ph8,0.025mnacl,1mm二亚乙基三胺五乙酸(dtpa))中的溶液中用三(2-羧乙基)膦(tcep,4.0摩尔当量)在37℃下处理2小时。在缓冲液(25mm硼酸钠ph8,0.025mnacl,1mmdtpa)中使用带有30kdanmwl的amiconultra-15离心过滤装置移除过量的tcep,接着用化合物7(20摩尔当量)在25℃下处理4小时。将反应混合物截断,并在ph7.4pbs缓冲液中使用带有30kdanmwl的amiconultra-15离心过滤装置浓缩,得到曲妥珠单抗-brd4-protac1。
实施例2:曲妥珠单抗-brd4-protac-2的制备
化合物8的合成
化合物8:向4-(n-顺丁烯二酰亚胺甲基)环己烷-1-甲酸丁二酰亚胺(smcc)(0.75mmol)在乙腈(7ml)中的溶液中添加1,2-乙二硫醇(0.82mmol)。将反应混合物在室温下搅拌3小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。将残余物通过柱层析用乙酸乙酯:己烷(3:2)纯化,得到白色固体化合物8(34.8%产率)。
化合物9的合成
化合物9:向化合物7(0.02mmol)在乙腈/二甲基甲酰胺(1:1,6ml)中的溶液中添加化合物8(0.04mmol)。将反应混合物在室温下搅拌16小时。反应完成后,反应混合物用二氯甲烷及水萃取。接着,有机层用盐水洗涤并经mgso4干燥。减压除去有机溶剂。通过柱层析用乙酸乙酯:己烷(3:2)纯化残余物,得到黄色固体化合物9(86.2%产率)。
曲妥珠单抗-brd4-protac2的偶联
向曲妥珠单抗1mg(5.0mg/ml)在缓冲液(50mm磷酸钾、50mm氯化钠、2mmedta;ph6.5)中的溶液中缓慢添加30当量的9(5mm在dmso中)。将反应混合物在37℃下搅拌18小时。在ph7.4pbs缓冲液中使用带有30kdanmwl的amiconultra-15离心过滤装置脱盐并浓缩抗体制备物,得到曲妥珠单抗-brd4-protac2。
如上所述,本发明的apc具有支链形式,其中,抗体-l2连接子与protac中的l1连接子连接。如上述实施例所示,l2连接子与l1连接子的连接不会改变protac的a结合物或b结合物,并且连接反应相对容易。作为比较,以下实施例说明合成直链(非支链)apc的尝试,其中,l2连接子与a结合物或b结合物偶联。
实施例3:直链形式的brd4-protac的合成(17)
图4说明可能的经由arv-825的a结合物连接合成的合成方案。蛋白质配体或连接酶结合物的官能团修饰并不容易。此外,并非所有蛋白质配体或连接酶结合物都具有合适的供修饰用的功能基团。在此实施例中,otx015(protacarv-825的蛋白质配体)上的氯原子难以转化为另一种官能团,如氨基。otx015或arv-825在布赫瓦尔德(buchwald)反应(钯催化的偶联反应)及乌尔曼(ullman)反应(铜催化的偶联反应)下显示无反应性。严苛的反应条件,如金属卤化物交换,将导致化合物分解。根据文献(ep1887008a1),应该在一开始就引入不同的brd4抑制剂官能团。换言之,将连接子与蛋白质配体或连接酶结合物直接偶联将使合成更复杂。
相反,本发明公开的支链连接子策略提供了连接任何蛋白质配体或连接酶结合物以形成具有以下优点的apc的新方法。apc保持靶蛋白配体及e3连接酶配体的结构,并因此结合亲和力不会改变。连接基团在连接子上的结构修饰比在配体上容易得多。连接子上的修饰适用于大多数protac,并且可以为不同的protac设计“共同的”偶联官能团,使得相同的抗体可以与不同的protac偶联,或者相同的protac可以与不同的抗体偶联。
实施例4:生物活性
测试各种式(i)的免疫偶联物降解靶向蛋白的特异性及能力。以下描述不同测定的简要描述。
western印迹(westernblot)
评估式(i)化合物在brd4蛋白质降解中的细胞效力。溴结构域蛋白4(brd4)是bet(溴结构域及额外)家族蛋白之一,并且涉及血液恶性肿瘤及实体肿瘤的肿瘤发生。brd4识别并结合乙酰化组蛋白,并在跨细胞分裂及转录调节的表观遗传记忆传递中起关键作用。靶向brd4的有效抑制剂显示出抗肿瘤活性,抑制各种癌细胞的增殖及转型。这导致brd4成为癌症治疗的有希望的治疗靶。已经显示brd4蛋白水解靶向嵌合体(protac)通过诱导brd4蛋白质降解而具有抗癌活性。然而,除了抗癌作用外,正常细胞也受这些药剂的影响。这导致了bet抑制的令人担忧的副作用,诸如自闭症样综合征及对记忆形成的损害。
在本发明中,产生具有支链形式的brd4-protac抗体偶联物,以保持brd4-protac模态的完整性,增强癌细胞靶向的特异性,并减少潜在的脱靶效应。“brd4-protac”是指包括brd4蛋白的靶结合物的protac。通过将“brd4-protac”与识别癌细胞上的抗原的抗体偶联。抗体的高特异性允许所得的偶联物(如ab-brd4-protac)靶向特定的癌细胞并对健康细胞产生较小的毒性。例如,抗体可以是曲妥珠单抗,而癌细胞可以是her2阳性bt-474乳腺癌细胞。在细胞brd4蛋白质降解中通过western印迹方法测试各种式(i)化合物(如,具有不同brd4结合物、不同e3结合物、及/或不同抗体)。以下使用曲妥珠单抗偶联的brd4-protac来说明本发明实施方式的益处。
对于western印迹实验,her2阳性bt-474及her2阴性mda-mb-231乳腺癌细胞分别在具有10%fbs的dmem及l15培养基中培养,并培养隔夜。在分析当天,用每种测试化合物预处理二十万个细胞24小时。24小时后,通过添加2×sds样品缓冲液收获全细胞裂解物。蛋白质通过sds-page电泳分离并转移至pvdf膜。根据标准方案使用各种一抗及二抗进行免疫印迹来检测蛋白质表达。抗brd4的抗体及抗兔igg、hrp连接的二抗购自cellsignalingtechnology公司(danvers,ma)。抗肌动蛋白的抗体购自millipore公司(burlington,ma)。免疫印迹通过化学发光(supersignaltmwestfemtomaximumsensitivitysubstrate,thermofisher公司,waltham,ma)显示,并通过chemidoctmmp成像系统(bio-rad公司,hercules,ca)检测。western印迹的条带强度也通过chemidoctmmp成像系统定量。将对应于药物处理组的条带的相对强度与未治疗组的相对强度进行比较。
图2显示分析结果。arv-825(cas号1818885-28-7)是异双功能分子,其包含与e3连接酶小脑蛋白结合部分连接的brd4结合部分。arv-825是蛋白水解靶向嵌合体(protac)。(参见lu,j.等人,“hijackingthee3ubiquitinligasecereblontoefficientlytargetbrd4”,chem.biol.22(6),755-763(2015))。本发明化合物5是arv-825的支链形式。本发明化合物7是具有溶酶体可切除的另外的二肽(缬氨酸-瓜氨酸)连接子的化合物5。
如图2所示,在此细胞培养分析中,化合物5与arv-825一样有效促进brd4的降解。相反,化合物7不能在高达1μm的化合物7处理时降解brd4蛋白质,此可能是由于缬氨酸-瓜氨酸二肽导致较低的渗透性所引起的。此结果表明,若apc在进入细胞之前过早地被切除,则释放的protac(例如,化合物7)将不会引起脱靶效应。因此,本发明的apc将具有更高的安全系数。
图3显示化合物7曲妥珠单抗抗体偶联物(如实施例1)在her2阳性bt-474乳腺癌细胞而非her2阴性mda-mb-231乳腺癌细胞中表达出特异性brd4蛋白降解活性。这种apc不会引起akt蛋白或肌动蛋白的降解。表明protac中抗体与连接子的支链偶联不会损害protac的活性。重要的是,在体内,抗体(曲妥珠单抗)将apc引导至表达特定抗原(例如her2)的那些细胞,借此减少脱靶效应。因此,实施例1在临床应用中与av-825一样有效但更安全。
这些结果表明,本发明的抗体偶联物(支链apc)可以增强癌细胞靶向的特异性,增强细胞对protac形式的摄取,并减少潜在的脱靶效应。另外,若抗体过早分离,则释放的protac(例如化合物7)由于缬氨酸-瓜氨酸二肽而具有相对较低的渗透性,并且不会引起不希望的脱靶效应,从而产生高安全裕度。
抗增殖活性
如上所述,本发明的apc可用于治疗携带特定抗原的疾病或失调。这些疾病可以是癌症、自体免疫疾病、感染性疾病、或血管增生性失调。癌症可能是肺癌、结肠癌、结直肠癌、乳腺癌、前列腺癌、肝癌、胰腺癌、膀胱癌、胃癌、肾癌、唾液腺癌、卵巢癌、子宫体癌、宫颈癌、口腔癌、皮肤癌、脑癌、淋巴瘤、或白血病。使用celltitertm-96分析法测量本发明的apc对细胞生长的抑制。在具有不同her2表达表型的乳腺癌细胞株中评估apc的细胞毒性。结果显示本发明的apc仅对her2阳性乳腺癌细胞有毒,其中,brd4蛋白、src激酶、或ras蛋白可通过使用与曲妥珠单抗偶联的合适protac而经特异性靶向以便蛋白酶体降解。
本发明的apc是有前景的新治疗剂,因为其具有adc及protac的优点。此外,本发明的支链apc显示优于直链adcproac,并且可用于治疗携带特定抗原的失调。
本发明的一些实施方式涉及使用本发明的apc治疗疾病或失调的方法。所述疾病可能是癌症。癌症的具体实例可包括乳腺癌、胃癌、鳞状细胞癌、结肠癌、及表达特定抗原的白血病。用于apc的抗体可以是曲妥珠单抗、西妥昔单抗(cetuximab)、利妥昔单抗(rituximab)、本妥昔单抗(brentuximab)、吉妥珠单抗(gemtuzumab)、伊珠单抗(inotuzumab)、萨希珠单抗(sacituzumab)、阿仑单抗(alemtuzumab)、尼妥珠单抗(nimotuzumab)。apc的具体实例可以是支链曲妥珠单抗偶联的protac,用于靶向具有her2表达的乳腺癌或胃癌。
支链抗体偶联的protac可以不同的形式合成,诸如不同的连接子或不同的抗体偶联方法。化合物18及化合物19是显示用于赖氨酸偶联的不同连接子形式的实施例。化合物18及化合物19中protac的合成遵循与具有peg连接子的化合物9相同的方法。化合物18及化合物19可以用与上述实施例2相同的方法合成。具有peg连接子的支链protac可以表现出更好的溶解度及与抗体的偶联。
用有限数量的实施例说明了本发明的实施方式。本领域技术人员将理解,在不脱离本发明的范围的情况下,其他修改及变化是可能的。因此,保护范围应仅受所附权利要求书的限制。
- 还没有人留言评论。精彩留言会获得点赞!