六方氮化硼的改性方法及羟基改性氮化硼与流程
本发明属于六方氮化硼技术领域,尤其涉及一种六方氮化硼的改性方法及羟基改性氮化硼。
背景技术:
六方氮化硼(h-bn)是石墨的等电子体,具有与石墨类似的层状结构,被称为“白色石墨”。除了具有类似石墨的优良润滑性、机械性能和导热性外,h-bn还具备许多独特的性能,例如绝缘性好(带隙宽度为5.2-5.8ev),热稳定性高(氮气气氛中使用温度可达2800℃)且不易氧化(氧化温度高于800℃),耐辐射以及生物相容性好(wengqh,wangxb,wangx,etal.chemicalsocietyreviews,2016,45(14),3989-4012;zhicy,bandoy,tangcc,etal.journalofmaterialschemistry,2008,18(33),3900-3908.)。近年来,用h-bn纳米管或纳米片作为填料改善聚合物基体的热学性能和绝缘阻燃性的研究引起了广泛的关注。研究发现,在环氧树脂中加入1wt%的bn纳米粒子(平均粒径2μm)便可使环氧树脂的释热速率峰值降低近一半(yub,xingw,guow,etal.journalofmaterialschemistrya,2016,4(19),7330-7340.);加入50wt%的bn纳米片(平均长度1.56μm,平均厚度2.35nm)可以使复合物热导率提高40倍(yujh,mohl,jiangpk.polymersforadvancedtechnologies,2015,26(5),514-520.)。
然而,h-bn化学性质稳定,表面缺乏活性基团,与大多数有机分子及聚合物分子链间缺乏强相互作用,因而在有机溶剂与聚合物基体中的分散性很差,这大大限制了h-bn对聚合物材料性能的改善效果。另一方面,h-bn的化学惰性又使其难以与其他物质反应进行表面改性。因此,开发经济、高效的h-bn表面修饰方法,增强其表面活性和反应性,是高性能聚合物/bn复合材料制备中一个关键且具有挑战性的课题。
h-bn的化学改性最早是利用h-bn表面的缺陷,使其与功能单体或聚合物反应,在表面引入烷基链(zhicy,bandoy,tangcc,etal.angewandtechemieinternationaledition,2005,44(48),7932-7935.),碳-卤键(zhicy,bandoy,tangcc,etal.journalofphysicalchemistryc,2007,111(3),1230-1233.)和聚甲基丙烯酸甲酯等高分子链(zhicy,bandoy,wangwl,etal.journalofnanomaterials,2008,1,145.),从而提高bn在溶剂中的分散性以及与聚合物的亲和性。但h-bn表面缺陷的密度和种类受限于其制备方法,难以控制。之后,人们提出采用适当的富电子试剂如过氧化氢、有机过氧化物、卡宾和萘钠体系等先进攻硼原子产生活性中心,再进行改性反应的方法(sainsburyt,o’neilla,passarellimk,etal.chemistryofmaterials,2014,26(24),7039-7050;shinh,guanj,zgierskimz,etal.acsnano,2015,9(12),12573-12582;zhicy,bandoy,teraot,etal.chemistry-anasianjournal,2009,4(10),1536-1540;sainsburyt,sattia,mayp,etal.journaloftheamericanchemicalsociety,2012,134(45),18758-18771.)。但由于b-n共价键十分稳定,即便是采用高活性的富电子试剂也需要高温或强氧化剂存在等严苛的条件,反应才能进行。最近,lei等将bn粉末与尿素粉在700rpm下球磨混合20小时(bn与尿素粉重量比为1:60),利用高剪切力作用和碰撞产生的高能量,得到2nm厚的bn纳米片,与此同时,还在bn表面引入氨基,大大提高了bn纳米粒子在水中的分散性,分散液浓度可以达到30mg/ml,较之前改性方法处理的bn纳米粒子的分散浓度提高了近30倍(leiww,mochalinvn,liud,etal.naturecommunications2015,6,8849.)。总的来说,在惰性的bn表面产生活性中心需要注入很高的能量,使现有的改性方法都涉及苛刻的反应条件,因此开发简单、高效又环保的bn改性方法对推动bn的应用意义重大。
技术实现要素:
有鉴于此,本发明的目的在于提供一种六方氮化硼的改性方法及羟基改性氮化硼,本发明提供的改性方法条件温和,易于操作,且得到的羟基改性氮化硼易于进一步改性。
本发明提供了一种六方氮化硼的改性方法,包括以下步骤:
将六方氮化硼粉末在硝酸中进行预处理,得到硝酸预处理的氮化硼;
将所述硝酸预处理的氮化硼分散在甲酸和硫酸的混合水溶液中,得到分散液;
将所述分散液进行γ射线辐照处理,得到羟基改性氮化硼。
本发明首先将六方氮化硼粉末在硝酸中进行预处理,然后将其分散在甲酸和硫酸的混合水溶液中,采用γ射线进行辐照处理,得到羟基改性氮化硼。本发明提供的方法条件温和,易于操作。
本发明首先将六方氮化硼粉末在硝酸中进行预处理,得到硝酸预处理的氮化硼。在一个实施例中,所述硝酸的质量分数为10%~30%。在一个实施例中,所述预处理的温度为80~90℃,预处理时间为5~8h。
所述预处理优选在搅拌的条件下进行,预处理完毕后将得到的预处理液离心分离,洗涤干燥后即可得到硝酸预处理的氮化硼。具体而言,所述预处理可以按照以下步骤操作:
称取h-bn粉末放于圆底烧瓶中,向其中加入硝酸溶液,并放入搅拌磁子。圆底烧瓶置于油浴中,开动磁力搅拌,加热并保持一定时间后,自然冷却至室温。将预处理液置于高速离心机中,以6000~10000rpm的转速,将硝酸处理的bn粒子离心分离,并用去离子水漂洗,至洗涤液呈中性。处理过的产物在40~80℃下干燥20~30h。
得到硝酸预处理的氮化硼后,将其分散在甲酸和硫酸的混合水溶液中,得到分散液,然后采用γ射线进行辐照处理,得到羟基改性氮化硼。在一个实施例中,所述分散液中,甲酸浓度为0.01~0.1mol/l,硫酸浓度为0.4mol/l,硝酸预处理的氮化硼的浓度为1~2mg/ml。
本发明优选在搅拌的条件下将硝酸预处理的氮化硼分散在甲酸和硫酸的混合水溶液中,并在搅拌的条件下对得到的分散液进行辐照处理,处理完毕后将得到的溶液静置使粒子沉积,将得到的粒子清洗、干燥即可获得羟基改性的氮化硼。具体而言,可以按照以下步骤处理:
将硝酸预处理的bn粉末放于圆底烧瓶中,加入去离子水,置于超声分散仪中使粉末超声分散均匀。将甲酸和浓硫酸依次加入其中,并放入搅拌磁子。圆底烧瓶用铁架台固定置于钴源室内,开动磁力搅拌,进行辐照,辐照结束后取出圆底烧瓶,静置一段时间后,使粒子沉积。将上层液体倒出,用去离子水漂洗沉淀物至洗涤液呈中性,洗净的沉淀物在40~80℃干燥10~20h,即得羟基改性bn粉末(ho-bn)。
在本发明的一个实施例中,采用60coγ射线对所述分散液进行辐照处理;所述辐照处理的吸收剂量率为80~120gy/min,总吸收剂量为100kgy以上。
得到羟基改性的氮化硼以后,还可以对其进行进一步改性,获得其他基团改性的氮化硼。
在一个实施例中,可以采用功能性硅氧烷对羟基改性的氮化硼进行进一步改性,步骤如下:
将所述羟基改性六方氮化硼分散在功能性硅氧烷溶液中,加热反应。
在本发明中,所述功能性硅氧烷为含有功能基团,例如环氧基、双键或氨基的硅氧烷,可以是市售的硅烷偶联剂,例如kh系列等。硅氧烷溶液中硅氧烷的体积分数为0.5%~3%,所述羟基改性氮化硼分散在功能性硅氧烷溶液中后浓度为1~4mg/ml;所述加热反应的温度为70~130℃,反应时间为7~10h。
本发明优选在搅拌的条件下使羟基改性的氮化硼和功能性硅氧烷反应,反应完毕后将产物洗涤、干燥,即可得到其他功能基团改性的氮化硼。具体而言,其可以按照以下步骤操作:
将ho-bn纳米粉置于三口烧瓶中,加入甲苯,置于超声分散仪中使粉末超声分散均匀,加入硅烷偶联剂,并放入搅拌磁子,在磁力搅拌下加热反应,反应结束后,静置使产物沉积。将上层液体倒出,用甲苯漂洗产物3次后,置于40~80℃下干燥10~20h,得到其他功能基团改性的bn纳米粉。
在一个实施例中,还可以采用反应单体对羟基改性的氮化硼进一步改性,步骤如下:
将所述羟基改性氮化硼分散在乙烯类单体溶液中,加入阻聚剂,进行第二次辐照处理,得到聚合物改性氮化硼。
在本发明中,所述乙烯类单体优选为极性乙烯类单体,包括但不限于丙烯酸、丙烯酸甲酯或甲基丙烯酸缩水甘油酯;所述第二次辐照的吸收剂量率为20~35gy/min,总吸收剂量为20~45kgy。
本发明优选在搅拌的条件下使羟基改性氮化硼与乙烯类单体进行反应,在搅拌的条件下进行第二次辐照,辐照结束后抽滤,干燥即可得到聚合物改性氮化硼。具体而言,其操作步骤如下:
将羟基改性bn纳米粉置于圆底烧瓶中,加入去离子水和cuso4·5h2o,将烧瓶置于超声分散仪中使粉末超声分散均匀加入乙烯类单体并放入搅拌磁子,体系通氮,封口,放入钴源室内,在磁力搅拌下辐照。辐照结束后,体系用孔径为0.22μm的纤维素微滤膜抽滤,所得滤饼重新分散在去离子水中,再抽滤,重复几次至滤液不再浑浊。将滤饼置于40~80℃下干燥10~20h。
本发明将六方氮化硼粉末在硝酸中进行预处理,然后将其分散在甲酸和硫酸的混合水溶液中,采用γ射线进行辐照处理,得到羟基改性氮化硼。本发明利用水溶液和bn的辐射化学效应,简单、高效的实现了h-bn纳米粒子的活性官能团改性,使其分散性和化学活性增加,扩大了其应用范围,为制备高性能高分子/bn纳米复合材料提供了可能。同其它改性方法相比,本方法在常温常压下直接利用γ射线辐射技术对h-bn进行简单高效的表面改性,具有条件温和、易于操作、环境友好和高效节能的特点。同时,该羟基改性bn纳米粉可继续分散于反应介质中,进一步得到其他活性官能团或聚合物改性的bn纳米粉。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1为硝酸处理前后h-bn的红外光谱;
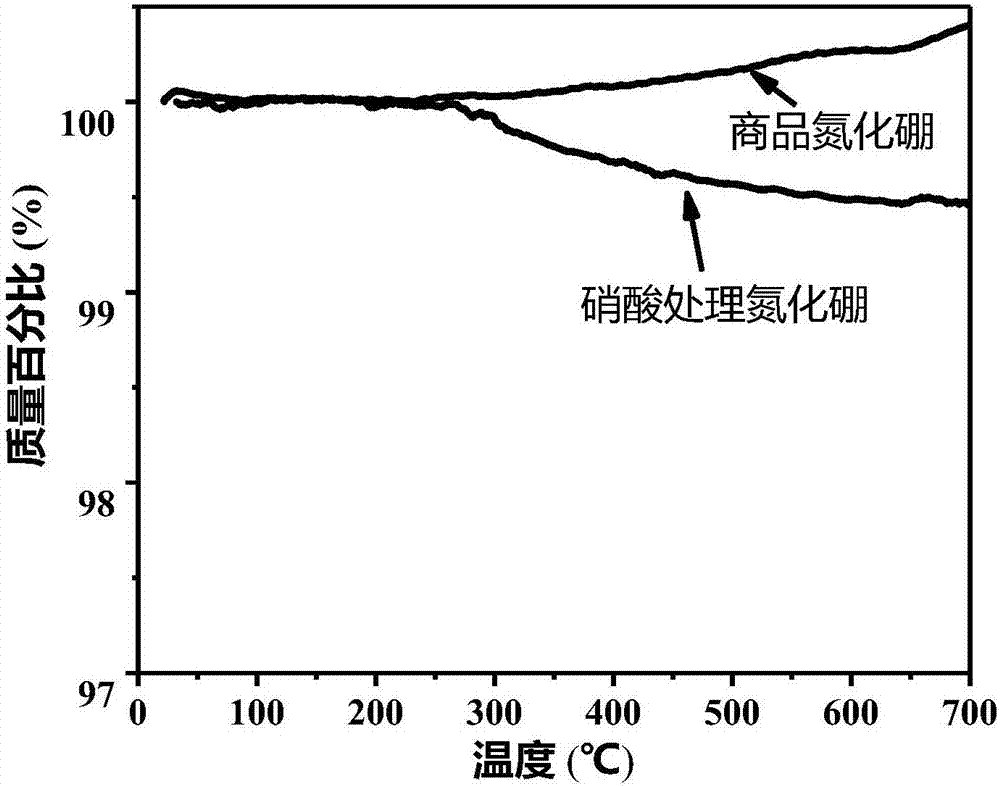
图2为硝酸处理前后h-bn在空气中加热的tg曲线;
图3为实施例1制备的ho-bn的红外光谱;
图4为实施例1制备的ho-bn在空气中加热的tg曲线;
图5为实施例2制备的ho-bn的红外光谱;
图6为实施例2制备的ho-bn在空气中加热的tg曲线;
图7为实施例3制备的ep-bn红外光谱;
图8为实施例3制备的ep-bn在空气中加热的tg曲线;
图9为实施例4制备的paa-bn的红外光谱;
图10是实施例4制备的paa-bn在空气中加热的tg曲线;
图11是本发明比较例1获得的产物的ft-ir光谱图;
图12是本发明比较例1获得的产物在空气中加热的tg曲线;
图13是本发明比较例2获得的产物的ft-ir光谱图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下实施例中,商品h-bn购于ge试剂公司,纯度≥99%,其余试剂均为分析纯。
实施例1羟基改性氮化硼(ho-bn)的制备
1)商品h-bn粉末预处理
称取2g商品h-bn粉末放于250ml圆底烧瓶中,向其中加入80ml10%的硝酸溶液(65%硝酸:水=1:5.5(v/v)),并放入搅拌磁子。圆底烧瓶置于油浴中,开动磁力搅拌,加热至80℃,保持5h后,自然冷却至室温,得到分散液。将分散液置于高速离心机(hc-2518)中,以8000rpm的转速,将bn粒子离心分离,并用去离子水漂洗,至洗涤液呈中性,处理过的产物在50℃电热鼓风干燥箱(gzx-9070mbe)中干燥24h。预处理前后bn粉末的ft-ir光谱如图1所示,图1为硝酸处理前后h-bn的红外光谱,由图1可知,两者基本没有差别,1395cm-1的吸收峰归属为b-n键的面内伸缩振动,804cm-1的吸收峰归属为b-n-b键的面外弯曲振动。但预处理前后bn粉末在空气中的热失重(tg)曲线如图2所示,图2为硝酸处理前后h-bn在空气中加热的tg曲线,由图2可知两者有较大差别,商品bn粉末中可能含有微量可氧化基团,导致在空气中加热时质量反而稍有增加。而经硝酸处理后的bn在空气中加热至300℃后稍有失重(约0.5%),说明硝酸可将bn上微量可氧化基团全部氧化。
2)bn粉末辐射羟基化改性
称取100mg硝酸处理后的bn粉末放于100ml圆底烧瓶中,加入50ml去离子水,置于超声分散仪中使粉末超声分散均匀。将20μl甲酸和1.1ml浓硫酸依次加入其中,并放入搅拌磁子。圆底烧瓶用铁架台固定置于钴源室内,开动磁力搅拌,辐照24h,吸收剂量率为113.4gy/min(硫酸亚铁剂量计标定),总吸收剂量为163.3kgy。辐照结束后取出圆底烧瓶,静置一段时间后,使粒子沉积。将上层液体倒出,用去离子水漂洗沉淀物至洗涤液呈中性。洗净的沉淀物在50℃电热鼓风干燥箱(gzx-9070mbe)中干燥24h,即得羟基改性bn粉末(ho-bn),其ftir光谱如图3所示,图3为实施例1制备的ho-bn的红外光谱,对比图1中辐照前bn的红外光谱,经辐照处理的bn在1155cm-1处出现新的吸收峰,这应归属于b-oh中b-o-h弯曲振动(kurapatir,backesc,ménard-moyonc,etal.angewandtechemie,2016,128(18),5596-5601.),说明bn上已经引入了-oh官能团。经辐照处理的bn在空气中加热的tg曲线如图4所示,图4为实施例1制备的ho-bn在空气中加热的tg曲线,图4显示加热至300℃时样品开始出现热失重,直至600℃至恒重,总失重率约为22%,而文献中用其他方法改性的bn失重率一般在5%以下(cuiz,oyeraj,gloveraj,schniepphc,adamsondh.small,2014,10(12),2352-2355;yub,xingw,guow,qius,wangx,los,huy.journalofmaterialschemistrya,2016,4(19),7330-7340.),此结果表明辐射法对bn的羟基改性程度较其他方法高得多。
实施例2羟基改性氮化硼(ho-bn)的制备
1)h-bn粉末预处理与实施例1相同。
2)ho-bn的制备
称取100mg硝酸处理后的h-bn粉末于100ml圆底烧瓶中,加入50ml去离子水,置于超声分散仪中使粉末超声分散均匀。再将20μl甲酸和1.1ml浓硫酸依次加入其中,并放入搅拌磁子。圆底烧瓶用铁架台固定置于钴源室内,开动磁力搅拌,辐照48h,吸收剂量率为105.4gy/min(硫酸亚铁剂量计标定),总吸收剂量为303.4kgy。辐照结束后取出圆底烧瓶,静置一段时间后,使粒子沉积。将上层液体倒出,用去离子水漂洗沉淀物至洗涤液呈中性。洗净的沉淀物在50℃电热鼓风干燥箱(gzx-9070mbe)中干燥24h,即得到ho-bn纳米粒子,其ftir谱图如图5所示,图5为实施例2制备的ho-bn的红外光谱,由图5可知,1155cm-1处出现b-o-h的特征吸收峰。空气中的tg曲线如图6所示,图6为实施例2制备的ho-bn在空气中加热的tg曲线,由图6可知,加热至400℃开始出现明显的热失重,直至600℃。总失重百分比约为38%。
实施例3环氧基团改性氮化硼(ep-bn)的制备
1)h-bn粉末预处理与实施例1相同。
2)ho-bn的制备
称取100mg硝酸处理后的h-bn粉末于100ml圆底烧瓶中,加入50ml去离子水,置于超声分散仪中使粉末超声分散均匀。再将20μl甲酸和1.1ml浓硫酸依次加入其中,并放入搅拌磁子。圆底烧瓶用铁架台固定置于钴源室内,开动磁力搅拌,辐照18h,吸收剂量率为101.3gy/min(硫酸亚铁剂量计标定),总吸收剂量为109kgy。辐照结束后取出圆底烧瓶,静置一段时间后,使粒子沉积。将上层液体倒出,用去离子水漂洗沉淀物至洗涤液呈中性。洗净的沉淀物在50℃电热鼓风干燥箱(gzx-9070mbe)中干燥24h,即得到ho-bn纳米粉末。
3)ep-bn的制备
称取80mg步骤2)制备的ho-bn纳米粉于50ml三口烧瓶中,加入20ml甲苯,置于超声分散仪中使粉末超声分散均匀。称取360mg硅烷偶联剂kh560加入上述分散液,并放入搅拌磁子。在磁力搅拌下加热至115℃,反应7h。反应结束后,静置使产物沉积。将上层液体倒出,用甲苯漂洗产物3次后,置于50℃电热鼓风干燥箱(gzx-9070mbe)中干燥12h,得到ep-bn纳米粉,ftir谱图如图7所示,图7为实施例3制备的ep-bn红外光谱。对比ho-bn的红外光谱(图3和图5),可以看出,在917cm-1和840cm-1左右出现新的吸收峰,这分别归属于环氧基团的环对称伸缩振动和环的面内变形振动,说明环氧基团已被引入bn纳米粒子上。ep-bn纳米粉在空气中加热的tg曲线如图8所示,图8为实施例3制备的ep-bn在空气中加热的tg曲线,可以看到有两个热失重区间,分别为270~400℃和400~600℃。对比ho-bn的tg曲线(图4和图6),270~400℃的热失重应对应于环氧基团的分解。
实施例4聚丙烯酸改性氮化硼纳米粒子(paa-bn)的制备
称取40mg实施例3中制备的ho-bn纳米粉置于50ml圆底烧瓶中。加入25ml去离子水和34.5mgcuso4·5h2o后,烧瓶置于超声分散仪中使粉末超声分散均匀。称取2.5ml精制丙烯酸加入分散液中,并放入搅拌磁子。体系通氮10min,封口,放入钴源室内。在磁力搅拌下辐照18h,吸收剂量率为22.8gy/min(硫酸亚铁剂量计标定),总吸收剂量为24.6kgy。辐照结束后,体系用孔径为0.22μm的纤维素微滤膜抽滤。所得滤饼重新分散在去离子水中,再抽滤。重复几次至滤液不再浑浊。将滤饼置于50℃电热鼓风干燥箱(gzx-9070mbe)中干燥12h,得到paa-bn,其ftir谱图见图9所示,图9为实施例4制备的paa-bn的红外光谱,由图9可知,在875cm-1和1795cm-1处出现b-c和c=o的特征吸收峰,说明bn上已接枝paa。paa-bn在空气中加热的tg曲线如图10所示,图10是实施例4制备的paa-bn在空气中加热的tg曲线,由图10可知,从220℃开始出现热失重,也说明paa-bn中有机碳骨架的存在。
比较例1
与实施例1操作步骤相同,区别在于,不对六方氮化硼进行硝酸预处理。所得产物的ft-ir光谱图见图11所示,图11是本发明比较例1获得的产物的ft-ir光谱图,其与原料氮化硼的红外光谱基本没有差别,并没有在1155cm-1处出现b-oh中b-o-h弯曲振动吸收峰。同时,该产物在空气中的热失重(tg)曲线见图12,图12是本发明比较例1获得的产物在空气中加热的tg曲线,其也没有明显的热失重。此结果表明,硝酸预处理的步骤对获得羟基改性氮化硼是必须的。
比较例2
与实施例1操作步骤相同,区别在于,硝酸改性的六方氮化硼分散在相同浓度的硫酸溶液中。所得产物的ft-ir光谱图见图13所示,图13是本发明比较例2获得的产物的ft-ir光谱图,其与原料氮化硼的红外光谱基本没有差别,并没有在1155cm-1处出现b-oh中b-o-h弯曲振动吸收峰。此结果表明,甲酸的存在对获得羟基改性氮化硼也是必须的。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!