一种氮化硼界面的制备方法与流程
本发明涉及一种氮化硼界面的制备方法,具体涉及在晶须或纤维表面氮化硼界面的制备。其主要应用于陶瓷基复合材料界面制备领域。
背景技术
碳化硅陶瓷基复合材料具有高比强度、高比模量、耐高温、抗氧化等一系列优异性能,可广泛应用于航空、航天领域。为了在更加严苛复杂的应力环境下服役,新兴的具有各向同性特征的碳化硅陶瓷基复合材料如碳化硅晶须(sicw)增韧陶瓷基复合材料、碳化硅短切纤维增韧陶瓷基复合材料等被开发出来。通常陶瓷基复合材料中增强体与基体间的界面结合较强,需要引入适当的弱结合界面来实现界面相载荷传递和力学熔断功能,最大限度地发挥增强体的增韧效果。选择合适的界面材料与界面制备工艺是保证复合材料性能优异的关键一环。
热解碳(pyc)和氮化硼(bn)能够同时满足低模量和底剪切强度要求,是常用的陶瓷基复合材料界面材料。热解碳(pyc)是目前最常用,制备技术最成熟的界面材料,能够在增强体与基体间形成适当的弱结合界面,更好的发挥增强体的增韧效果。但热解碳(pyc)在低温下易氧化,使所制备的陶瓷基复合材料的抗氧化性下降,限制了陶瓷基复合材料的应用范围。氮化硼具有类石墨结构,热稳定性好,介电性能优良;较之于热解碳(pyc),氮化硼(bn)抗氧化性能更好,且高温氧化环境下的产物为液态氧化硼(b2o3),一方面可以有效阻止氧的扩散,另一方面液态氧化硼(b2o3)还可以填补材料内部的微裂纹起到一定的自愈合效果。所以,氮化硼(bn)是具有较大发展潜力的界面材料。
现有的复合材料界面制备工艺多针对传统的具有各向异性特征的连续纤维增韧陶瓷基复合材料。这种具有各向异性特征的陶瓷基复合材料的预制体大多具有两级孔隙结构(大于100微米的大孔隙与小于10微米的小孔隙),该结构有利于界面的渗透,使预制体的增强体表面形成厚度均匀连续的界面。但是具有各向同性特征的碳化硅陶瓷基复合材料的预制体内部为单极小孔隙结构(小于10微米的小孔隙),该结构不利于界面的渗透,使现有的界面制备工艺很难在预制体的增强体表面形成厚度均匀连续的界面。因此,在这种具有单极小孔隙结构预制体的增强体表面制备厚度均匀连续的界面成为目前亟需解决的问题。
典型的氮化硼(bn)界面制备方法有三种:化学气相渗透法(cvi)、浸渍-涂覆法(dip-coating)、先驱体浸渍裂解法(pip)。化学气相渗透法多采用bcl3-nh3反应体系,当预制体内部为多级孔隙时,有利于反应气体的扩散,氮化硼(bn)界面的渗透能力较强,可在预制体内部形成厚度均匀的界面;当预制体内部为单极孔隙且孔隙尺寸较小时,反应气体扩散较为困难,氮化硼(bn)界面的渗透能力较弱,造成预制体内部形成的界面厚度不均匀。谢亚南等在文献“界面对sicw/sic层状陶瓷性能的影响”中采用cvi法在sicw/sic(sicw为sic晶须)层状陶瓷层内制备bn界面后,层状陶瓷的强度和韧性分别下降了15.38%和25.05%,这是由于cvi-bn是以扩散传质为主,气态前驱体的进入和扩散慢于等压热梯度cvi,sicw/sic层状陶瓷的层内为单极小孔隙,气体输送速度慢于沉积反应速度,使层内的bn界面厚度出现梯度,降低了材料的机械性能。浸渍-涂覆法(dip-coating)多采用硼酸-尿素反应体系,该法将硼酸与尿素以一定比例溶于乙醇-水混合溶液中,然后将制得的溶液浸渍预制体,经干燥后在氮气或氨气气氛下高温反应得到氮化硼(bn)界面。由于采用液相浸渍的方法引入硼源和氮源,因此浸渍-涂覆法适用于孔隙尺寸较小厚度较大的预制体。另外,该工艺具有操作简单,成本低并且反应副产物无毒的优点,但这种方法存在界面不连续,质量不均匀,转化率低等缺点。liu等在文献“mechanicalandmicrowavedielectricpropertiesofsicf/siccompositeswithbninterphasepreparedbydip-coatingprocess”中,使用浸渍-涂覆法在sic纤维表面制备氮化硼(bn)界面,所制备的sicf/bn/sic复合材料的弯曲强度是未制备氮化硼(bn)界面sicf/sic复合材料的两倍,但存在sic纤维表面bn界面不连续,浸渍-涂覆过程中的热处理造成sic纤维损伤等不足。先驱体浸渍裂解法(pip)将含有硼元素和氮元素的有机先驱体溶于有机溶剂中,然后将制得的溶液浸渍预制体,经干燥和高温裂解得到氮化硼(bn)界面。先驱体浸渍裂解法(pip)同样适用于孔隙尺寸较小厚度较大的预制体,但所制备的氮化硼(bn)界面具有较多的裂纹与气孔。
因此,采用上述的工艺方法在具有单极小孔隙的预制体中制备氮化硼(bn)界面存在较多缺陷。化学气相渗透法(cvi)虽然制备的氮化硼(bn)界面连续且致密,但氮化硼(bn)界面的渗透性有限,若预制体孔隙较小或厚度较大,则易在预制体内部形成厚度不均匀的界面。浸渍-涂覆法(dip-coating)与先驱体浸渍裂解法(pip)采用的是液相浸渍法引入硼源和氮源,氮化硼(bn)界面的制备受预制体厚度的影响小,但也存在界面不连续,晶化程度与转化率较低等一系列问题。
技术实现要素:
要解决的技术问题
为了避免现有技术的不足之处,本发明提出一种氮化硼界面的制备方法,解决单极小孔隙的预制体中制备氮化硼(bn)界面困难,涂层质量不高的问题。
技术方案
一种氮化硼界面的制备方法,其特征在于步骤如下:
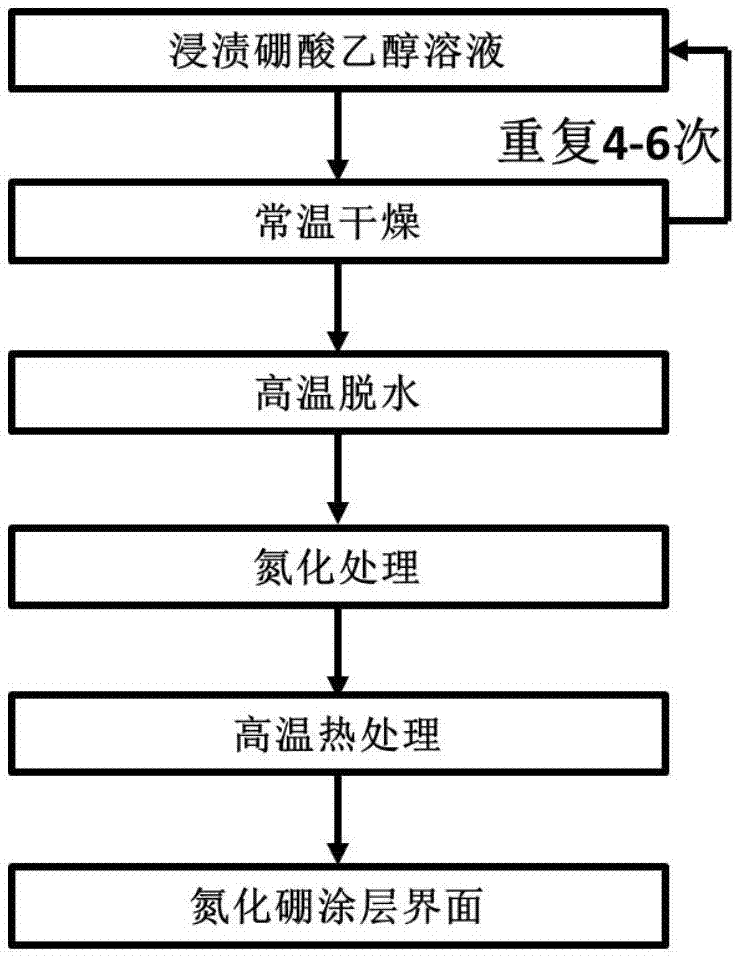
步骤1、浸渍硼酸乙醇溶液与常温干燥:将预制体放入真空浸渍皿中,抽真空20~30min,然后将预制体放入硼酸乙醇溶液中,继续抽真空20~30min,将预制体取出,在常温下干燥12h后,继续完成该步骤3~6次;
所述硼酸乙醇溶液的摩尔分数为0.5~2mol/l,硼酸为溶质,乙醇为溶剂;
步骤2、高温脱水:将步骤1的预制体放入烘箱,在100~110℃下保温1~2h,再升温至150~180℃,保温3~5h,完成高温脱水;
步骤3、氮化处理:将步骤2的预制体放入气氛炉等温区,对预制体进行氮化处理,氮化温度:900~1100℃,保温时间:8~16h,炉内压力:500~2000pa,气氛:nh3-h2,利用以下反应得到氮化硼界面:b2o3+2nh3→2bn+3h2o;
步骤4、高温热处理:将步骤3得到的预制体放入热处理炉内高温热处理,热处理温度1400~1800℃,保温时间0.5~2h,炉内压力:100~200kpa,气氛:n2,提高氮化硼界面的晶化程度并除去残余的氧化硼。
所述氮化处理的气氛:nh3-h2,nh3-h2的比例为1﹕2。
所述步骤4中的高温热处理的气压为常压或高压。
所述预制体为:陶瓷晶须或陶瓷纤维。
所述陶瓷晶须为非氧化晶须sic晶须或氧化物晶须al2o3晶须。
陶瓷纤维为非氧化纤维sic纤维或氧化物纤维al2o3纤维。
有益效果
本发明提出的一种氮化硼界面的制备方法,在预制体的增强体表面制备厚度均匀、连续并且质量稳定的氮化硼(bn)界面的制备方法。本发明的思想在于将硼酸乙醇溶液浸渍预制体,经过常温干燥,高温脱水,然后在低压氨气氛(氨气和氢气混合)中氮化处理,最后经高温热处理得到厚度均匀、连续并且质量稳定的氮化硼(bn)界面。
本发明的有益效果有以下几点:
(1)所用的硼源为前期引入,氮化处理时的气氛(nh3-h2)是小分子,在低压下的渗透能力强,较之于化学气相渗透法(cvi),可以在孔隙更小厚度更大的预制体中制备氮化硼界面,并且可在预制体内部形成厚度均匀的界面,这种制备方法既适用于各向同性复合材料,也适用于各向异性复合材料。
(2)采用两步法先后引入硼源和氮源,低压通入氨气与氧化硼缓慢反应,较之于浸渍-涂覆法(dip-coating),反应更加温和,使预制体的增强体表面的界面连续均匀。另外,在浸渍过程中,只需配制硼酸乙醇溶液,不用考虑硼源与氮源在溶液内的配比,因此可以引入更多的硼源,从而提高氮化硼界面的转化率。
整个工艺过程操作简单,可重复性高,成本低廉,可用于制备各向同性或各向异性的大尺寸复杂构件。
附图说明
图1.是本发明的工艺流程图。
图2.是本发明实施例1sic晶须预制体内部制备的氮化硼界面断面扫描电子显微镜(sem)照片。
图3.是本发明实施例1sic晶须预制体内部制备氮化硼界面的拉曼光谱。
图4.是本发明实施例1sic晶须预制体内部制备氮化硼界面的红外光谱。
具体实施方式
现结合实施例、附图对本发明作进一步描述:
实施例1.
步骤一:制备sic晶须预制体。
步骤二:浸渍硼酸乙醇溶液与常温干燥。配置摩尔分数为1mol/l的硼酸乙醇溶液,将配好的溶液放入真空浸渍皿准备浸渍。将sic晶须预制体放入真空浸渍皿,抽真空20min,然后将sic晶须预制体放入溶液中,继续抽真空20min,将sic晶须预制体取出,在常温下干燥12h后,重复完成该步骤3次。
步骤三:高温脱水。干燥后将sic晶须预制体放入烘箱,在110℃下保温2h,再升温至180℃,保温5h,完成高温脱水。
步骤四:氮化处理。将步骤三的sic晶须预制体放入气氛炉等温区,对预制体进行氮化处理,氮化温度:950℃,保温时间:8h,炉内压力:1000pa,气氛:nh3-h2,利用以下反应得到氮化硼界面:b2o3(s)+2nh3(g)→2bn(s)+3h2o(g)。
步骤五:高温热处理。将步骤四得到的sic晶须预制体放入热处理炉内高温热处理,热处理温度1700℃,保温时间1h,炉内压力:100kpa,气氛:n2,提高氮化硼界面的晶化程度并除去残余的氧化硼。
实施例2.
步骤一:制备sic短切纤维毡预制体(sic纤维商品代号:h-nicalons)。
步骤二:浸渍硼酸乙醇溶液与常温干燥。配制摩尔分数为1.5mol/l的硼酸乙醇溶液,将配好的溶液放入真空浸渍皿准备浸渍。将sic短切纤维毡预制体放入真空浸渍皿,抽真空30min,然后将sic短切纤维毡预制体放入溶液中,继续抽真空30min,将sic短切纤维毡预制体取出,在常温下干燥12h后,重复完成该步骤3次。
步骤三:高温脱水。干燥后将sic短切纤维毡预制体放入烘箱,在110℃下保温2h,再升温至150℃,保温4h,完成高温脱水。
步骤四:氮化处理。将步骤三的sic短切纤维毡预制体放入气氛炉等温区,对预制体进行氮化处理,氮化温度:1100℃,保温时间:6h,炉内压力:1000pa,气氛:nh3-h2,利用以下反应得到氮化硼界面:b2o3(s)+2nh3(g)→2bn(s)+3h2o(g)。
步骤五:高温热处理。将步骤四得到的sic短切纤维毡预制体放入热处理炉内高温热处理,热处理温度1400℃,保温时间1h,炉内压力:150kpa,气氛:n2,提高氮化硼界面的晶化程度并除去残余的氧化硼。
实施例3.
步骤一:制备单向sic纤维预制体(sic纤维商品代号:tyrannosa)。
步骤二:浸渍硼酸乙醇溶液与常温干燥。配制摩尔分数为1.5mol/l的硼酸乙醇溶液,将配好的溶液放入真空浸渍皿准备浸渍。将单向sic纤维预制体放入真空浸渍皿,抽真空30min,然后将单向sic纤维预制体放入溶液中,继续抽真空30min,将sic纤维预制体取出,在常温下干燥12h后,重复完成该步骤5次。
步骤三:高温脱水。干燥后将单向sic纤维预制体放入烘箱,在110℃下保温2h,再升温至180℃,保温4h,完成高温脱水。
步骤四:氮化处理。将步骤二的单向sic纤维预制体放入气氛炉等温区,对单向sic纤维预制体进行氮化处理,氮化温度:1000℃,保温时间:6h,炉内压力:1000pa,气氛:nh3-h2,利用以下反应得到氮化硼界面:b2o3(s)+2nh3(g)→2bn(s)+3h2o(g)。
步骤五:高温热处理。将步骤四得到的单向sic纤维预制体放入热处理炉内高温热处理,热处理温度1400℃,保温时间1h,炉内压力:150kpa,气氛:n2,提高氮化硼界面的晶化程度以及除去残余的氧化硼。
实施例4.
步骤一:制备2.5维或3维sic纤维预制体(sic纤维商品代号:sylramic)。
步骤一:浸渍硼酸乙醇溶液与常温干燥。配制摩尔分数为1.5mol/l的硼酸乙醇溶液,将配好的溶液放入真空浸渍皿准备浸渍。将sic纤维预制体放入真空浸渍皿,抽真空30min,然后将sic纤维预制体放入溶液中,继续抽真空30min,将sic纤维预制体取出,在常温下干燥12h后,重复完成该步骤5次。
步骤二:高温脱水。干燥后将sic纤维预制体放入烘箱,在110℃下保温2h,再升温至180℃,保温4h,完成高温脱水。
步骤三:氮化处理。将步骤二的sic纤维预制体放入气氛炉等温区,对sic纤维预制体进行氮化处理,氮化温度:900℃,保温时间:6h,炉内压力:800pa,气氛:nh3-h2,利用以下反应得到氮化硼界面涂层:b2o3(s)+2nh3(g)→2bn(s)+3h2o(g)。
步骤四:高温热处理。将步骤四得到的sic纤维预制体放入热处理炉内高温热处理,热处理温度1400℃,保温时间1h,炉内压力:200kpa,气氛:n2,提高氮化硼界面的晶化程度并除去残余的氧化硼。
图2sic晶须预制体内部制备的氮化硼界面断面扫描电子显微镜(sem)照片;(a)为采用本发明制备氮化硼界面后预制体内部形貌的低倍扫描照片;(b)(c)为采用本发明制备氮化硼界面后预制体内部形貌的高倍扫描照片;(d)为采用dip-coating法制备氮化硼界面后预制体内部形貌的高倍扫描照片。
图3sic晶须预制体内部制备氮化硼界面的拉曼光谱;a0为sicw预制体的拉曼光谱,a1为采用cvi法制备氮化硼界面后的预制体的拉曼光谱,a2采用本发明制备氮化硼界面后的预制体的拉曼光谱,a3采用dip-coating法制备氮化硼界面后的预制体的拉曼光谱。
图4sic晶须预制体内部制备氮化硼界面的红外光谱;s0为sicw预制体的红外光谱,s1为采用cvi法制备氮化硼界面后的预制体的红外光谱,s2采用本发明制备氮化硼界面后的预制体的红外光谱。
- 还没有人留言评论。精彩留言会获得点赞!