腐胺生产能力提高的重组微生物及用其制备腐胺的方法与流程
本发明涉及生产腐胺的能力提高的重组微生物,以及利用所述微生物生产腐胺的方法。
背景技术:
腐胺(或1,4-丁二胺)是如亚精胺和精胺等的一类聚胺,存在于革兰氏阴性细菌和真菌中。由于在各种物种中腐胺存在的浓度范围很大,因此估计其在微生物代谢中起着重要作用。腐胺通常采用化学合成的方法,通过源于丙烯的琥珀腈和丙烯腈生产。所述化学合成采用衍生自石油化学品的物质作为起始原料,还利用有毒的化学物质,因此,该方法不是环境友好的而且具有石油消耗的问题。为了解决这些问题,很多研究试图开发利用微生物来生物合成腐胺的方法,这种方法对环境更友好并减少能源消耗。根据现有的知识,可通过两种微生物途径生物合成腐胺。一种途径是,谷氨酸盐产生鸟氨酸,而鸟氨酸脱羧基合成腐胺。另一种途径是,鸟氨酸合成精氨酸,通过精氨酸产生胍丁胺,然后再从胍丁胺合成腐胺。此外,其他合成腐胺的方法利用转化有参与已知腐胺合成途径的酶的目标微生物。例如,WO09/125924公开了高收率生产腐胺的方法,该方法使大肠杆菌(E.coli)中参与腐胺分解和利用的途径失活、使作为腐胺前体的鸟氨酸转化为精氨酸的途径失活、和增强鸟氨酸的生物合成途径。一篇在2010年出版的文献公开了一种生产高浓度腐胺的方法,所述方法将使鸟氨酸转化为腐胺的蛋白导入不产腐胺的棒状杆菌(Corynebacterium)菌株,并提高其活性。此外,还公开了从精氨酸产生腐胺的方法,该方法将源自大肠杆菌的精氨酸脱羧酶和胍丁胺酶(agmatinase)引入菌株。就此而言,鸟氨酸途径的腐胺产量是精氨酸途径的约50倍(Schneider等.,Appl.Microbiol.Biotechnol.88:4,859-868,2010)。
技术实现要素:
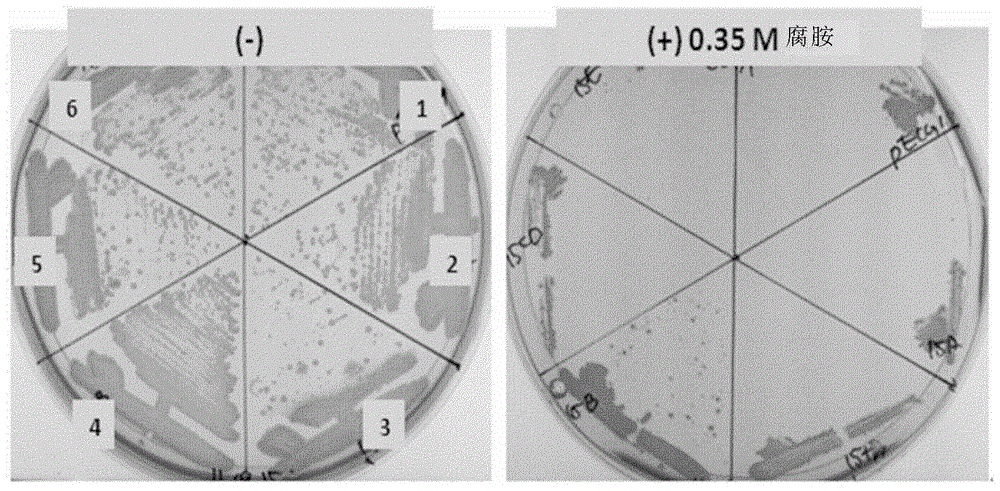
技术问题在此背景中,本发明人发现,通过弱化或除去NCgl0101蛋白的活性,可以在棒状杆菌属的微生物中高收率地生产腐胺,从而完成本发明。技术方案本发明的目的之一是提供能高收率地生产腐胺的棒状杆菌属的重组微生物,所述微生物经修饰而具有相比于内源活性得到弱化的NCgl0101活性。本发明的另一目的是提供利用该微生物产生腐胺的方法。有益效果当利用产腐胺能力提高的本发明棒状杆菌属微生物来产生腐胺时,其经修饰,从而与内源活性相比,弱化了NCg10101活性,因此,其能高收率地产生腐胺。因此,所述微生物可广泛用于更有效地产生腐胺。附图说明图1是显示编码NCgl0100、NCgl0101、NCgl0102、NCgl0103和NCgl0104的基因在野生型谷氨酸棒状杆菌ATCC13032菌株的染色体(chromosome)上相对位置的示意图;和图2显示了在本发明制备的重组菌株之间作生长比较的测试结果,其中,1、2、3、4、5和6是将pHC139T、pHC139T-P(CJ7)-NCgl0100、pHC139T-P(CJ7)-tNCgl0100、pHC139T-P(CJ7)-NCgl0101、pHC139T-P(CJ7)-NCgl0102-NCgl0103和pHC139T-P(CJ7)-NCgl0104分别引入KCCM11138P而制备的菌株。最佳方式为实现上述目的,在一方面,本发明提供产生腐胺能力提高的棒状杆菌属重组微生物,与内源活性相比,所述微生物经弱化或除去NCg10101蛋白的活性而得以修饰,所述蛋白具有SEQIDNO.17或SEQIDNO.19所示氨基酸序列。本文所用的术语“NCg10101”表示显示出金属依赖酶活性的蛋白,其在谷氨酸棒状杆菌中表达,其功能尚未完全了解。其包括肽酶M20家族或氨基苯甲酰基-谷氨酸利用蛋白(AbgB)的金属结合域或氨基丙氧基。大肠杆菌的AbgB构成含AbgB的氨基苯甲酰基-谷氨酸水解酶,将氨基苯甲酰基-谷氨酸水解成氨基苯甲酸盐和谷氨酸盐。氨基苯甲酸盐已知用作叶酸合成的前体,但其与腐胺产生的关系尚未知。本发明的NCgl0101蛋白可包含SEQIDNO:17或SEQIDNO:19所示氨基酸序列。然而,不限于此,因为根据微生物种类或菌株,该蛋白的氨基酸序列可能有所不同。换言之,其可以是突变型蛋白或人工变体,所述突变型蛋白或人工变体的氨基酸序列在SEQIDNO:17或SEQIDNO:19所示氨基酸序列的一个或多个位置包含一个或几个氨基酸的取代、缺失、插入或添加,只要有助于通过弱化该蛋白的活性而提高产腐胺能力。本文的“几个”可根据蛋白中氨基酸残基的三维结构的位置或类型而有所不同,但尤其表示2-20,特别是2-10,更特别是2-5。此外,根据微生物的个体或种类,氨基酸的取代、缺失、插入、添加或倒置包括人工变体或天然突变所致的那些。编码本发明氨基酸序列的多核苷酸可包括编码以下蛋白的多核苷酸序列:具有SEQIDNO:17或SEQIDNO:19所示氨基酸序列的蛋白,或者其氨基酸序列与SEQIDNO:17或SEQIDNO:19所示氨基酸序列有80%或更高、特别是90%或更高、更特别是95%或更高、尤其是97%或更高同源性的蛋白,只要该蛋白具有与NCg10101蛋白相似的活性。最特别的是SEQIDNO:16或SEQIDNO:18所示多核苷酸序列。术语“同源性”表示两条氨基酸序列之间的同一性,其可采用本领域技术人员熟知的方法检测,采用BLAST2.0来计算诸如评分、同一性和相似性等参数。此外,本发明的编码NCg10101的多核苷酸序列可与SEQIDNO:16所示多核苷酸或由其制备的探针在“严格条件”下杂交,也可以是编码能正常起作用的NCg10101蛋白的经修饰的多核苷酸。本文所用的“严格条件”是指允许多核苷酸间进行特异性杂交的条件,具体描述于,例如,《分子克隆》(实验室手册,J.Sambrook等编,第二版,冷泉港实验室出版,纽约冷泉港,1989)或《最新分子生物学方法》(F.M.Ausubel等编,JohnWiley&Sons公司,纽约)。例如,在65℃的杂交缓冲液中进行杂交(3.5×ssc,0.02%Ficoll,0.02%聚乙烯吡咯烷酮,0.02%牛血清白蛋白,2.5mMNaH2PO4(pH7),0.5%SDS,2mMEDTA)。SSC是pH为7的0.15M氯化钠/0.15M柠檬酸钠。杂交之后,在室温下用2×SSC清洗转移了DNA的膜,然后在68℃下采用0.1-0.5×ssc/0.1×SDS进行清洗。可通过以下方式弱化本发明NCg10101蛋白的活性:1)编码该蛋白的多核苷酸的部分或完全缺失,2)修饰表达调控序列以降低该多核苷酸的表达,3)修饰染色体上的序列或4)它们的组合。在上文中,可利用染色体基因插入的载体,通过将编码内源性靶蛋白的多核苷酸替换为标记基因或部分核苷酸序列缺失的多核苷酸来实施编码蛋白的多核苷酸的部分或完全缺失。“部分”缺失的长度可根据多核苷酸的种类而有所不同,但尤其是2bp-300bp,更特别是2bp-100bp,更特别是1bp-5bp。还可通过以下方式修饰表达调控序列来降低多核苷酸表达:通过核苷酸序列的缺失、插入、保守或非保守性取代或它们的组合在表达调控序列中诱导突变以进一步弱化表达调控序列的活性,或将表达调控序列替换成活性更低的序列。表达调控序列包括编码启动子的序列、操纵子序列、核糖体结合位点和控制转录和翻译终止的序列。此外,可通过以下方式修饰染色体上的多核苷酸序列以弱化蛋白的活性:通过核苷酸序列的缺失、插入、保守或非保守性取代或它们的组合在序列中诱导突变以进一步弱化该序列的活性,或将多核苷酸序列替换成经修饰的序列以便获得更弱的蛋白活性。同时,还可进一步修饰产腐胺能力增强的本发明棒状杆菌属微生物,从而相比于内源活性,弱化参与鸟氨酸合成精氨酸的鸟氨酸氨甲酰基转移酶(ArgF)的活性和参与谷氨酸输出的蛋白(NCg10101)的活性。此外,可通过额外导入鸟氨酸脱羧酶(ODC)活性来修饰棒状杆菌属微生物。相较于内源性活性,还可进一步修饰棒状杆菌属微生物以增强乙酰谷氨酸合酶将谷氨酸转化成乙酰谷氨酸的活性,增强鸟氨酸乙酰转移酶(ArgJ)将乙酰鸟氨酸转化为鸟氨酸的活性,增强乙酰谷氨酸激酶(ArgB)将乙酰谷氨酸转化为乙酰谷氨酰磷酸的活性,增强乙酰γ谷氨酰磷酸还原酶(ArgC)将乙酰谷氨酰磷酸转化为乙酰谷氨酸半醛的活性,和增强乙酰鸟氨酸氨基转移酶(ArgD)将乙酰谷氨酸半醛转化为乙酰鸟氨酸的活性,藉此增强作为腐胺前体的鸟氨酸的生物合成途径(SakanyanV等.,Microbiology.142:1,99-108,1996)。在此例中,ArgF、NCg11221、ODC、ArgC、ArgJ、ArgB和ArgD可分别具有(但不特定限于)SEQIDNO.:20、21、22、23、24、25、26所示的氨基酸序列,或与这些序列有80%或更高、特别是90%或更高、更特别是95%或更高,尤其是97%或更高的同源性的氨基酸序列。本文所用的术语“鸟氨酸脱羧酶(ODC)”是指利用鸟氨酸产生腐胺的酶,ODC需要磷酸吡哆醛(吡哆醛5'-磷酸,PLP)作为辅酶。ODC见于大部分革兰氏阴性菌,也可见于一些肠道细菌,例如革兰氏阳性细菌-乳酸菌。大肠杆菌有两种编码ODC的基因,其一为speC,在特定浓度下持续表达,而另一种speF则在特定条件(在有高于一定浓度的鸟氨酸存在下以及低pH下)下表达。根据物种不同,某些物种,如大肠杆菌有两种ODC,而其他仅有一种。诸如埃希氏菌属(Escherichiasp.)、志贺氏菌属(Shigellasp.)、枸橼酸杆菌属(Citrobactersp.)、沙门氏菌属(Salmonellasp.)和肠杆菌属(Enterobactersp.)等物种有两种ODC(speC,speF),而耶尔森式菌属(Yersiniasp.)、克雷白氏菌属(Klebsiellasp.)、欧文氏菌属(Erwiniasp.)等菌株则有一种ODC(speC)。在乳酸菌中,ODC由一种基因(speF)表达,其可以在低pH值或充足的鸟氨酸和组氨酸的条件下诱导表达。可利用源自多个不同物种的ODC编码基因将ODC的活性引入本发明的棒状杆菌属重组微生物。编码ODC的多核苷酸可包括(但不限于)编码由以下氨基酸序列组成的蛋白的多核苷酸:SEQIDNO.:22所示氨基酸序列,或者与该氨基酸序列有70%或更高、特别是80%或更高,更特别是90%或更高同源性的氨基酸序列。此外,可通过本领域熟知的各种方法将鸟氨酸脱羧酶(ODC)活性引入微生物;例如,将包含ODC编码核苷酸序列的多核苷酸插入染色体的方法,通过引入载体系统而将多核苷酸导入微生物的方法,将经修饰或活性增强的启动子插入ODC编码核苷酸序列上游的方法,和对ODC编码核苷酸序列插入突变的方法。更特别是,如果引入了ODC的编码核苷酸序列,可利用已知的CJ7启动子作为启动子来控制其表达。此外,可通过以下方式实现ArgC、ArgJ、ArgB和ArgD活性的增强:1)增加编码酶的多核苷酸拷贝数;2)修饰表达调控序列以提高多核苷酸的表达;3)修饰染色体上酶的编码多核苷酸序列以增强所述酶的活性;或4)以上的组合。在方法1)中,可通过将多核苷酸操作性连接于载体,或将其插入宿主细胞的染色体来实现编码酶的多核苷酸拷贝数增加。更具体地,可通过引入能够独立复制并行使功能的载体来增加宿主细胞的多核苷酸拷贝数,其中,编码本发明酶的多核苷酸操作性相连;或可通过引入能够将多核苷酸插入宿主细胞染色体的载体来增加宿主细胞的多核苷酸拷贝数,其中所述多核苷酸操作性相连。本文所用的术语“载体”是指含有靶蛋白编码多核苷酸的核苷酸序列的DNA构建物,其操作性连接于合适的调控序列以在合适的宿主中表达靶蛋白。调控序列包括引发转录的启动子、控制转录的任何操纵序列、编码合适的mRNA核糖体结合位点的序列和控制转录和翻译终止的序列。所述载体可转染入适合的宿主,然后独立于宿主基因组进行复制和行使功能,其自身可整合入染色体。在本发明中,可利用本领域已知的任何载体,而无任何特殊限制,只要其能在宿主中复制。常用载体的例子是自然状态或重组状态下的质粒、黏粒、病毒和噬菌体。例如,pWE15、M13、λMBL3、λMBL4、λIXII、λASHII、λAPII、λt10、λt11、Charon4A和Charon21A可用作噬菌体载体或黏粒载体,而pBR系统、pUC系统、pBluescriptII系统、pGEM系统、pTZ系统、pCL系统和pET系统可用作质粒载体。可用于本发明的载体没有特殊限制,可利用已知的表达载体。具体地说,可利用pACYC177、pACYC184、pCL、pECCG117、pUC19、pBR322、pMW118、pCC1BAC载体。更具体是,可利用pACYC177、pCL、pCC1BAC载体。此外,可将编码靶蛋白的多核苷酸插入宿主细胞染色体的载体可以具体是,例如,穿梭载体pECCG112(韩国专利公开号No.1992-0000933),其能在大肠杆菌和棒状菌(Coryneformbacteria)中自我复制,但不限于此。此外,可利用染色体基因插入的载体将染色体中靶蛋白的编码多核苷酸替换成新的多核苷酸。可通过本领域任何已知的方法实现多核苷酸插入染色体,例如,通过同源重组。由于可通过诱导同源重组将本发明载体插入染色体,可额外包含选择标记来确认基因成功插入染色体。选择标记用于筛选转化有载体的细胞,换言之,为了确定目标多核苷酸是否插入。可利用可提供可选择表型的标记,所述表型是例如药物抗性、营养缺陷型、对有毒物质的耐受性或表面蛋白的表达。在用选择性物质处理的环境中,只有表达选择性标记的细胞可以存活,或者细胞显示不同表型,因此可通过这种方法来选择成功转化的细胞。如本文所用,术语“转化”是指将含有靶蛋白的编码多核苷酸的载体引入宿主细胞,从而该蛋白可以在该细胞中表达。转化的多核苷酸包括编码靶蛋白的所有多核苷酸,其能在宿主细胞中表达而无论其定位,是插入宿主细胞的染色体还是定位在染色体外。此外,所述多核苷酸包括编码靶蛋白的DNA和RNA。可引入任何形式的多核苷酸,只要其能引入宿主细胞并表达。例如,可采用表达盒的形式(基因构建物)将多核苷酸引入宿主细胞,其含有自我表达所需的所有必须元件。所述表达盒通常包括操作性连接于多核苷酸的启动子、转录终止信号、核糖体结合位点和翻译终止信号。表达盒可以是能够自我复制的表达载体的形式。此外,多核苷酸可以其自身的形式引入宿主细胞,并操作性连接于宿主细胞表达所需的序列。本文所用的术语“操作性连接”是指在启动或介导编码靶蛋白的多核苷酸转录的启动子序列与所述多核苷酸之间的功能性连接。此外,可如下实施方法2),修饰表达调控序列以增强本发明多核苷酸的表达:通过核苷酸序列的缺失、插入、保守或非保守性取代、或其组合在序列中引入突变,或替换成活性增强的核苷酸序列。所述表达调控序列包括启动子、操纵序列(operatorsequence)、核糖体结合位点编码序列和控制转录和翻译终止的序列。可将强异源启动子连接于多核苷酸表达单元(unit)的上游以取代原始启动子。强启动子的例子有pcj7启动子、lysCP1启动子、EF-Tu启动子、groEL启动子、aceA或aceB启动子等,更具体是,操作性连接源自棒状杆菌的lysCP1启动子或pcj7启动子以增强酶的编码多核苷酸表达。在本文中,lysCP1启动子是通过天冬氨酸激酶和天冬氨酸半醛脱氢酶的编码多核苷酸的启动子区域核苷酸序列取代获得的增强启动子,通过增强天冬氨酸激酶基因的表达,其足够强从而将相应酶的活性相比于野生型提高了5倍(国际申请公开号2009-096689)。此外,鉴定到pcj7启动子在产氨棒状杆菌(Corynebacteriumammoniagenes)和埃希氏菌(Escherichia)中表达并具有强启动子活性,其在谷氨酸棒状杆菌(Corynebacteriumglutamicum)中也能高强度表达(韩国专利号0620092)。此外,可如下所述(但不特别限于此)实施方法3),修饰染色体上的多核苷酸序列:通过核酸序列的缺失、插入、保守或非保守性替换或它们的组合来诱导序列突变以增强所述序列的活性,或通过活性增强的核苷酸序列作取代。本发明的微生物是具有产腐胺能力的微生物,包括原核微生物,其中表达包含SEQIDNO:17或SEQIDNO:19所示氨基酸序列的蛋白,其可以是,例如埃希氏菌属(Escherichiasp.)、志贺氏菌属(Shigellasp.)、枸橼酸杆菌属(Citrobactersp.)、沙门氏菌属(Salmonellasp.)、肠杆菌属(Enterobactersp.)、耶尔森式菌属(Yersiniasp.)、克雷白氏菌属(Klebsiellasp.)、欧文氏菌属(Erwiniasp.)、棒状杆菌属(Corynebacteriumsp.)、短杆菌属(Brevibacteriumsp.)、乳杆菌属(Lactobacillussp.)、单胞菌属(Sllenomanassp.)和弧菌属(Vibriosp.)。本发明微生物具体是棒状杆菌(Corynebacterium)属微生物,更具体是谷氨酸棒状杆菌(Corynebacteriumglutamicum)。在本发明的一实施方式中,保藏号为KCCM11138P的棒状杆菌属微生物(韩国专利公开号No.2012-0064046)得到修饰,其通过增强腐胺生物合成途径而具有了产生高浓度腐胺的能力。具体地,产腐胺的菌株KCCM11138P是过度产生腐胺的菌株,其中,菌株ATCC13032缺失编码鸟氨酸氨甲酰基转移酶(ArgF)的基因以便促进鸟氨酸累积和编码谷氨酸转运蛋白(glutamateexporter,NCgl1221)的基因以便增加细胞内谷氨酸,引入鸟氨酸脱羧酶(speC)的编码基因,并增加鸟氨酸生物合成基因(argCJBD)的表达水平。在本发明的另一实施方式中,修饰了基于谷氨酸棒状杆菌ATCC13869的腐胺产生菌株DAB12-a。该菌株ATCC13869基于与KCCM11138P相同的基因型,KCCM11138P是基于谷氨酸棒状杆菌ATCC13032的腐胺产生菌株。具体地说,腐胺产生菌株DAB12-a来自从美国模式培养物保藏中心(ATCC)获得的ATCC13869菌株,其中缺失鸟氨酸氨甲酰基转移酶(ArgF)的编码基因、编码蛋白NCgl1221以外运谷氨酸的基因,引入了源自大肠杆菌的鸟氨酸脱羧酶(ODC)编码基因(speC),而鸟氨酸生物合成基因操纵子(argCJBD)的启动子替换为增强的启动子。根据本发明的一实施方式,棒状杆菌属微生物(KCCM11138P)具有产腐胺的能力,该微生物通过以下方式制备:缺失鸟氨酸氨甲酰基转移酶(ArgF)的编码基因和参与谷氨酸外运的谷氨酸转运蛋白(NCgl1221)的编码基因,替换编码参与谷氨酸合成鸟氨酸的酶的ArgCJBD基因簇自身的启动子,和将鸟氨酸脱羧酶(ODC)的编码基因(speC)引入野生型谷氨酸棒状杆菌ATCC13032的染色体。基于KCCM11138P,选出在含有高浓度腐胺的培养基上生长良好的一克隆(A15),证实选出的A15包含编码NCgl0100、NCgl0101、NCgl0102、NCgl0103和NCgl0104的基因(实施例1)。此外,由于5类基因中编码NCg10101的基因,微生物在含有高浓度腐胺的培养基中生长(实施例2)。就NCg10101编码基因的特征而言,证实在编码NCg10101的基因过表达的菌株中腐胺产生降低(实施例3),在编码NCg10101的基因缺失的菌株中腐胺产生升高(实施例4)。因此,本发明人通过除去产生腐胺的菌株KCCM11138P中的NCg10101基因而制备得到产腐胺能力提高的谷氨酸棒状杆菌菌株,将该菌株命名为谷氨酸棒状杆菌CC01-0244,保藏于韩国微生物保藏中心(下文简写为“KCCM”),保藏日期2011年12月26日,登录号KCCM11241P。为实现以上目的,在本发明的另一方面,本发明涉及生产腐胺的方法,包括以下步骤:培养产腐胺能力提高的棒状杆菌属微生物,该微生物经修饰以具有弱化的NCg10101蛋白活性,所述NCg10101蛋白具有SEQIDNO.17或SEQIDNO.19所示的氨基酸序列;和从以上步骤所得的培养液中分离腐胺。本发明的培养方法可在本领域已知的合适培养基和培养条件下实施。本领域技术人员可以根据所选的菌株方便地调整和采用培养方法。培养方法的例子包括(但不限于)分批、连续和补料分批培养。培养基必须适当地满足具体菌株的要求。培养基必须适当地满足具体菌株的要求。用于各种微生物的培养基描述于,例如,《通用细菌学方法手册》(ManualofMethodsforGeneralBacteriology),美国细菌协会(AmericanSocietyforBacteriology),(华盛顿,哥伦比亚特区,美国,1981)。可利用以下作为培养基的碳源:糖和碳水化合物(例如,葡萄糖、蔗糖、乳糖、果糖、麦芽糖、糖蜜、淀粉和纤维素),乳脂和脂肪(例如,大豆油、葵花籽油、花生油和椰油),脂肪酸(例如,棕榈酸、硬脂酸和亚油酸),醇类(例如,丙三醇和乙醇)以及有机酸(例如,乙酸)等。这些物质可单独或混合使用。可利用以下作为氮源:含氮有机化合物(例如,蛋白胨,酵母提取物,牛肉提取物,麦芽提取物,玉米浆,豆粕粉和尿素)或非有机化合物(例如,硫酸铵,氯化铵,磷酸铵,碳酸铵和硝酸铵),而这些物质也可以单独或混合使用。可利用以下物质作为磷源:磷酸二氢钾或磷酸氢二钾或其相应的含钠盐。此外,培养基还可包括生长必要的金属盐(例如,硫酸镁或硫酸铁),最后,除了上述物质外,还可利用必要的生长促进物质,例如氨基酸和维生素。除培养基外,也可以加入合适的前体。进料物质可以在培养基中一次性提供或在培养过程中足量提供。可通过合适的碱性化合物(例如,氢氧化钠、氢氧化钾或氨)或酸性化合物(例如磷酸或硫酸)调节培养液的pH。可采用消泡剂(例如脂肪酸聚乙二醇酯)来调节起泡情况。可引入氧气或含氧气体混合物,例如空气来维持培养的需氧环境。培养温度通常为20-45℃,特别是25-40℃。可以持续培养直至腐胺产生达到所需上限,通常在10-160小时内达到该目标。腐胺可释放入培养基,或包含在细胞中。就收集和回收在本发明培养方法中产生的腐胺的方法而言,可根据培养方法(例如,分批、连续或补料分批培养),采用本领域已知的合适方法从培养基中回收目标物质。本发明的实施方式下文将参照以下实施例更详细地描述本发明。然而,这些实施例仅作为举例目的,本发明不应局限于这些实施例。实施例1:用于选择腐胺生物合成的有效基因的文库制备和克隆筛选为从野生型棒状杆菌菌株的染色体中筛选腐胺生物合成的有效基因,制备野生型棒状杆菌菌株的染色体文库。详细而言,用限制性酶Sau3AI随机切割从野生型谷氨酸棒状杆菌ATCC13032菌株提取的染色体,从中选择5-8kb的片段,然后克隆入大肠杆菌穿梭载体pECCG122(韩国专利公布号1992-0000933)以制备染色体文库。为从如此制备的棒状杆菌染色体文库中选择有效的腐胺生物合成基因,获得在含有高浓度腐胺的培养基中生长的菌落。同时,将这些文库引入具有产腐胺能力的棒状杆菌属微生物(KCCM11138P),从而制备各转化体。选择能在含有0.35M腐胺(10g/l葡萄糖、0.4g/lMgSO4·7H2O、4g/lNH4Cl、1g/lKH2PO4、1g/lK2HPO4、2g/l脲、10mg/lFeSO4·7H2O、1mg/lMnSO4·5H2O、5mg/l烟酰胺、5mg/l盐酸硫胺素、0.1mg/l生物素、1mM精氨酸、25mg/l卡那霉素、0.35M腐胺,pH7.0)的基本培养基中生长的转化体。菌株KCCM11138P在本发明人申请的专利(韩国专利公布号2012-0064046)中披露,该菌株通过以下方式制备:缺失野生型谷氨酸棒状杆菌菌株ATCC13032的染色体中编码鸟氨酸氨甲酰基转移酶(ArgF)和谷氨酸转运蛋白(NCgl1221)的基因,将源自大肠杆菌W3110菌株的鸟氨酸脱羧酶(ODC)的编码基因(speC)引入染色体,和替换编码参与谷氨酸合成鸟氨酸的酶的argCJBD基因簇的启动子,从而制备各转化体。结果为,选择了275个菌落,随后鉴定在含有高浓度腐胺的培养基中生长良好的菌落。获得各文库克隆并重新引入(产)腐胺菌株。随后,鉴定在含有高浓度腐胺的培养基中生长良好的菌落,因而最终选择了克隆(A15)。通过测序鉴定该选择的克隆。结果为,证实该克隆包含NCgl0100、NCgl0101、NCgl0102、NCgl0103和NCgl0104的总共5个ORF,其中除去了N-末端的436个氨基酸(图1)。图1是显示NCgl0100、NCgl0101、NCgl0102、NCgl0103和NCgl0104的编码基因的相对位置的示意图,这些基因在野生型谷氨酸棒状杆菌ATCC13032菌株的染色体上。实施例2:鉴定A15克隆中腐胺合成的有效基因实施例2-1:克隆A15克隆的5个基因和制备转化体实施例1所得A15克隆的核苷酸序列已经知晓。基于以前报道的ATCC13032菌株的核苷酸序列,构建SEQIDNO.1和2所示的NCgl0100-F和NCgl0100-R作为扩增NCgl0100基因的引物,SEQIDNO.2和3所示的NCgl0100-R和tNCgl0100-F作为扩增tNCgl0100基因的引物(其中除去了N-末端的436个氨基酸),SEQIDNO.4和5所示的NCgl0101-F和NCgl0101-R作为扩增NCgl0101基因的引物,SEQIDNO.6和7所示的NCgl0102-F和NCgl0103-R作为扩增NCgl0102和NCgl0103基因的引物,和SEQIDNO.8和9所示的NCgl0104-F和NCgl0104-R作为扩增NCgl0104基因的引物。此外,构建SEQIDNO.10和11所示的P(CJ7)-F和P(CJ7)-R作为扩增表达启动子P(CJ7)(或pcj7)(韩国专利号10-0620092)的引物(表1)。随后,利用ATCC13032菌株的染色体作为模板和SEQIDNO.1-9所示的各引物进行PCR(95℃下变性30秒,50℃下退火30秒,72℃下延伸1分钟~1分30秒,25轮),从而扩增5类基因片段。此外,利用产氨棒状杆菌的染色体作为模板和SEQIDNO.10和11所示的引物进行PCR以扩增启动子片段。将KpnI和XbaI切割的5种基因和EcoRV和KpnI切割的CJ7启动子连接入EcoRV和XbaI切割的表达载体pHC139T(韩国专利号10-0860932),从而制备总共5类表达载体,pHC139T-P(CJ7)-NCgl0100、pHC139T-P(CJ7)-tNCgl0100、pHC139T-P(CJ7)-NCgl0101、pHC139T-P(CJ7)-NCgl0102-NCgl0103和pHC139T-P(CJ7)-NCgl0104。表1制备表达A15克隆所含5种基因的菌株的引物NCgl0100-F(SEQIDNO.1)GCGCATATGAGCTCAACAACCTCAAAAACCNCgl0100-R(SEQIDNO.2)GCGTCTAGATTATCCTTCGAGGAAGATCGCAGtNCgt0100-F(SEQIDNO.3)GCGCATATGTGGACGCTGATGGCTGCNCgl0101-F(SEQIDNO.4)GCGCATATGAGTACTGACAATTTTTCTCCACNCgl0101-R(SEQIDNO.5)GCGTCTAGACTAAGCCAAATAGTCCCCTACNCgl0102-F(SEQIDNO.6)GCGCATATGGATGAACGAAGCCGGTTTGNCgl0103-R(SEQIDNO.7)GCGTCTAGATTAATCAATGAAGACGAATACAATTCCNCgl0104-F(SEQIDNO.8)GCGCATATGGCGGGTGACAAATTGTGGNCgl0104-R(SEQIDNO.9)GCGTCTAGATTAGGACAGTTCCGCTGGAGCP(CJ7)-F(SEQIDNO.10)CAGATATCGCCGGCATAGCCTACCGATGP(CJ7)-R(SEQIDNO.11)GCGTCTAGAGATATCAGTGTTTCCTTTCG通过电穿孔将如此制得到的5类表达载体和对照组pHC139T引入实施例1的KCCM11138P菌株,然后接种于含有25μg/ml卡那霉素的BHIS平板以选择转化体。实施例2-2:找寻腐胺的有效基因采用于实施例1相同的方式从实施例2-1获得的总共6类转化体中选择在含有高浓度腐胺的培养基中良好生长的转化体(图2)。图2是在本发明所制备转化体之间比较生长情况的测试结果,其中1、2、3、4、5和6表示分别引入6类以下表达载体的菌株:pHC139T、pHC139T-P(CJ7)-NCgl0100、pHC139T-P(CJ7)-tNCgl0100、pHC139T-P(CJ7)-NCgl0101、pHC139T-P(CJ7)-NCgl0102-NCgl0103和pHC139T-P(CJ7)-NCgl0104。如图2所示,仅有引入了pHC139T-P(CJ7)-NCgl0101的转化体(4号)在含有高浓度腐胺的培养基中显示生长良好,因此选择NCg10101作为腐胺生物合成的有效基因。实施例3:评估NCgl0101-过表达菌株产生腐胺的能力评估过表达NCg10101基因的菌株产腐胺的能力,该基因在实施例2中鉴定为有效基因。将pHC139T-P(CJ7)-NCgl0101引入产腐胺菌株KCCM11138P来制备用于评估的菌株。通过电穿孔将实施例2-1制备的pHC139T-P(CJ7)-NCgl0101和作为对照组的pHC139T载体引入产腐胺菌株KCCM11138P,然后接种于含有25μg/ml卡那霉素的BHIS平板以选择转化体。这些转化体分别命名为KCCM11138P/pHC139T和KCCM11138P/pHC139T-P(CJ7)-NCgl0101。30℃下,将如此选择的这两种转化体在含有1mM精氨酸(1%葡萄糖、1%聚蛋白胨、0.5%酵母提取物、0.5%牛肉提取物、0.25%NaCl、0.2%脲、100μl50%NaOH、2%琼脂、pH6.8,每1L)的CM平板上培养24小时,然后将一接种环的细胞培养物接种在25ml表2所示含有25μg/ml卡那霉素的滴定培养基,30℃下以200rpm振荡培养96小时。发酵期间,在培养基中加入1mM精氨酸培养所有制备的菌株。表2组成浓度(每1L)葡萄糖8%大豆蛋白0.25%玉米浆固体0.5%(NH4)2SO44%脲0.15%KH2PO40.1%MgSO47H2O0.05%生物素100μg盐酸硫胺素3000μg钙-泛酸3000μg烟酰胺3000μgCaCO35%结果如表3所示,当NCg10101过表达时,腐胺产量降低。表3菌株类型腐胺(g/L)KCCM11138P/pHC139T9.5KCCM11138P/pHC139T-P(CJ7)-NCgl01015.1实施例4:评估NCg10101-缺失菌株产生腐胺的能力实施例4-1:用基于ATCC13032的腐胺产生菌株制备NCg10101缺失菌株根据实施例3,NCg10101过表达增加细胞在含有高浓度腐胺的培养基中生长,但降低腐胺产量。基于此结果,检验NCg10101缺失对产腐胺能力的影响。详细地说,基于ATCC13032菌株的NCg10101核苷酸序列,构建SEQIDNO.12和13所示NCgl0101-del-F1_BamHI和NCgl0101-del-R1_SalI作为引物来获得NCg10101的N-末端区域同源重组片段。构建SEQIDNO.14和15所示NCgl0101-del-F2_SalI和NCgl0101-del-R2_XbaI作为引物来获得NCg10101的C-末端区域同源重组片段(表4)。采用这两对引物,通过PCR制备NCg10101基因的N-末端和C-末端区域的片段。用BamHI&SalI和SalI&XbaI分别处理PCR产物,克隆入BamHI&XbaI处理的pDZ载体。克隆的质粒命名为pDZ-NCgl0101(K/O)。表4制备NCgl0101-缺失菌株的引物NCgl0101-del-F1_BamHI(SEQIDNO.12)CGGGATCCCGGATTCCCTGCGATCATTGNCgl0101-del-R1_SalI(SEQIDNO.13)ACGCGTCGACCAGTCGACGGAACTTGTGGAGNCgl0101-del-F2_SalI(SEQIDNO.14)ACGCGTCGACGGCAACGACTCCGAAACCTTCNCgl0101-del-R2_XbaI(SEQIDNO.15)CTAGTCTAGACTGGATCCTCATGAATGCGC通过电穿孔将为获得KCCM11138PΔNCgl0101菌株制备的pDZ-NCgl0101(K/O)载体引入KCCM11138P菌株,然后接种于含有25μg/ml卡那霉素的BHIS平板。通过观察含有X-gal(5-溴-4-氯-3-吲哚基-β-D-半乳糖苷)的固体培养基上的菌落是否为蓝色来证实载体成功插入染色体。原代染色体插入菌株在营养培养基中振荡培养(30℃,8小时),然后从10-4稀释到10-10,接种于含X-gal的固体培养基。虽然大多数菌落显示为蓝色菌落,但少部分的菌落显示为白色菌落。最终筛选出了NCg10101基因-缺失的菌株,通过双交换的白色菌落,利用SEQIDNO.12和15所示引物,通过PCR鉴定。如此鉴定的变体命名为KCCM11138PΔNCgl0101。实施例4-2:用基于ATCC13869的腐胺产生菌株制备NCg10101缺失菌株利用基于谷氨酸棒状杆菌ATCC13869的腐胺产生菌株DAB12-a(argF-缺失、NCgl1221-缺失、引入大肠杆菌speC和arg操纵子-argCJBD启动子-取代的菌株)制备NCg10101缺失菌株,前者与基于谷氨酸棒状杆菌ATCC13032的产腐胺菌株KCCM11138P具有相同基因型。详细地说,为鉴定源自谷氨酸棒状杆菌ATCC13869的编码NCg10101的基因和其所表达蛋白的氨基酸序列,利用谷氨酸棒状杆菌ATCC13869的基因组DNA作为模板和一对引物SEQIDNO.12和15(NCgl0101-del-F1_BamHI、NCgl0101-del-R2_XbaI)进行PCR。本实施例中,PCR反应进行30轮:95℃下变性30秒,53℃下退火30秒,72℃下延伸2分钟30秒。通过电泳分离PCR产物,并分析它们的序列。经序列分析,鉴定到源自谷氨酸棒状杆菌ATCC13869的编码NCg10101的基因包含SEQIDNO.18所示核苷酸序列,其编码的蛋白包含SEQIDNO.19所示氨基酸序列。比较源自谷氨酸棒状杆菌ATCC13032的NCg10101的氨基酸序列与源自谷氨酸棒状杆菌ATCC13869的NCg10101的氨基酸序列时,二者显示有98%的序列同源性。为缺失源自谷氨酸棒状杆菌ATCC13869的NCg10101的编码基因,采用谷氨酸棒状杆菌ATCC13869的基因组DNA作为模板和表4所列的两对引物,以实施例4-1相同的方式,通过PCR扩增NCg10101基因的N-末端和C-末端区域。然后,用BamHI&SalI和SalI&XbaI分别处理PCR产物,克隆入BamHI&XbaI处理的pDZ载体,藉此构建质粒pDZ-2’NCgl0101(K/O)。采用与实施例4-1相同的方式将质粒pDZ-2’NCgl0101(K/O)转化入谷氨酸棒状杆菌DAB12-a,选择NCg10101编码基因缺失的菌株。选择的谷氨酸棒状杆菌变体命名为DAB12-aΔNCgl0101。实施例4-3:评估NCg10101-缺失菌株产生腐胺的能力为研究NCg10101缺失对产腐胺菌株产生腐胺能力的影响,比较实施例4-1和4-2中制备的谷氨酸棒状杆菌变体。详细地说,采用与实施例3相同的方式评估两类谷氨酸棒状杆菌变体(KCCM11138PΔNCgl0101和DAB12-aΔNCgl0101)产腐胺的能力。如下表5所示,发现NCg10101缺失提高了腐胺产量。表5菌株类型腐胺(g/L)KCCM11138P9.8KCCM11138PΔNCgl010111.3DAB12-a10.1DAB12-aΔNCgl010111.0综合来看,实施例3和4的结果显示,在野生型谷氨酸棒状杆菌菌株中,NCg10101编码基因过表达降低腐胺产量,而该基因缺失增加了腐胺产量,表明NCg10101直接影响腐胺生物合成。因此,在上述实施例中本发明人通过缺失产生腐胺的菌株KCCM11138P中的NCg10101基因而制备得到产腐胺能力提高的谷氨酸棒状杆菌菌株,将该菌株命名为谷氨酸棒状杆菌CC01-0244,并在2011年12月26日根据布达佩斯条约保藏于作为国际保藏机构的韩国微生物保藏中心(下文简写为“KCCM”),登录号KCCM11241P。基于以上描述,本领域技术人员应理解,可采用其他形式实施本发明,而不改变技术理念或必要技术特征。就此而言,上述实施例用于说明本发明的各方面,但不想要限制本发明的范围。应理解,本发明的范围包括从以下权利要求的含义、范围和等同概念所衍生的任何改变和改进形式,而非以上的具体描述。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1