一种硬脑/脊膜生物补片材料及其制备方法和应用与流程
本发明属于组织工程学医用生物材料技术领域,具体涉及一种硬脑/脊膜生物补片材料及其制备方法和应用。
背景技术:
硬脑膜和硬脊膜分别是大脑与颅骨、脊柱与脊髓之间重要的功能膜组织,具有相同的生物学特征和功能。硬脑膜贴附颅腔内面,与颅盖连结疏松,与颅底结合紧密;硬脑膜在脑神经颅处向外延伸,移行而与神经的被膜结合,在枕骨大孔周缘向下延为硬脊膜。硬脑膜与硬脊膜的功能和结构是一致的,硬脑膜用于保护脑组织,硬脊膜用于保护脊髓组织。外伤、肿瘤、炎症、神经外科手术或其他脑颅脊柱疾病等均会造成硬脑/脊膜缺损。硬脑/脊膜的缺损会引起癫痫、脊液外漏、颅内感染、脑膜膨出和脑功能障碍等病症。如在颅底外科手术中,颅底硬脑膜相对薄且与颅底骨结合紧密,一旦出现缺损易造成脑脊液鼻漏和耳漏。颅底的脑膜瘤、脊索瘤等肿瘤常常侵蚀浸润到周围的颅骨和硬脑膜,在切除肿瘤手术时,切除的肿瘤通常也会附带部分脑膜和颅骨,造成术后发生脑脊液漏和颅内感染。为治疗硬脑/脊膜缺损,现有技术中常采用硬脑/脊膜替代材料植入缺损部分进行修复硬脑/脊膜缺损。随着医学技术的高速发展,硬脑膜替代材料越来越广泛的应用于脑凸面手术、创伤直接损伤、肿瘤的浸润、手术过程中硬脑膜切开减压以及一些先天性的因素造成的硬脑膜缺损等诸多方面,如在脑外科开颅手术患者中有10%~15%需要硬脑/脊膜替代材料修复硬脑膜缺损,也有报道称该比例已经高达30%。因此,为了满足临床需要,开展研究合适的硬脑/脊膜修复材料至关重要。但是,现有的硬脑/脊膜替代材料在使用的过程中往往存在诸多不足,常常引起严重的并发症问题。例如在硬脑/脊膜替代材料在植入体内后降解速度控制方面的问题,若硬脑/脊膜替代材料降解的速度过快,撕裂力下降,则导致防液体渗漏作用下降,引起硬脑/脊膜与周围组织粘连、感染、出血、脑脊液漏以及癫痫等病症;但是若硬脑/脊膜替代材料长时间不降解,则又导致其与组织再生不匹配,生物相容性差,更甚者易导致炎症反应、结缔组织增生包裹、感染及出血等问题。因此,对于硬脑/脊膜替代材料在植入人体或动物体内的降解速度方面的控制是非常重要的。然而,在以往以天然膜材料生产硬脑/脊膜替代材料的工艺中,当膜材料经过冻干辐照后,天然膜材料中的胶原蛋白会被大幅破坏,使膜材料撕裂力下降,以及为了能够更好脱去细胞和脂肪,使用的脱细胞、抗原和脂肪的试剂对膜的破坏相对较大,使天然膜材料的其他力学性能大幅降低,最终导致其降解速度加快,防液体渗漏作用下降。为了降低生物组织修复材料的降解速度,现有技术中通常采用交联剂处理膜材料,提高其力学性能,降低其降解速度,使其在人体或动物体内能够长时间不降解。但是对于用于修复硬脑/脊膜缺损的替代材料来说,由于其特殊的生理位置以及功能决定了现有技术中用于处理其他部位的组织修复材料或硬脑/脊膜缺损的替代材料中的交联剂并不适用于硬脑/脊膜补片材料,主要存在的问题包括:(1)交联剂处理后,修复材料在人体或动物体内长时间不降解,导致其在植入人体或动物体内后,生物相容性差,易产生不利的副作用;(2)交联剂的稳定性低,大多数交联剂都具有比较容易反应的基团,这种基团导致交联剂不稳定,进而导致修复材料在人体或动物体内短时间内即降解,导致修复材料防液体渗漏作用下降,进而引发一系列术后并发症,带来不利的影响;(3)交联剂毒性高,难以控制交联剂最终在产品上的残留量,影响被植入的人或动物的身体健康。因此,现有技术中亟需开发一种更为适用于硬脑/脊膜修补降解性能要求的生物补片材料。
技术实现要素:
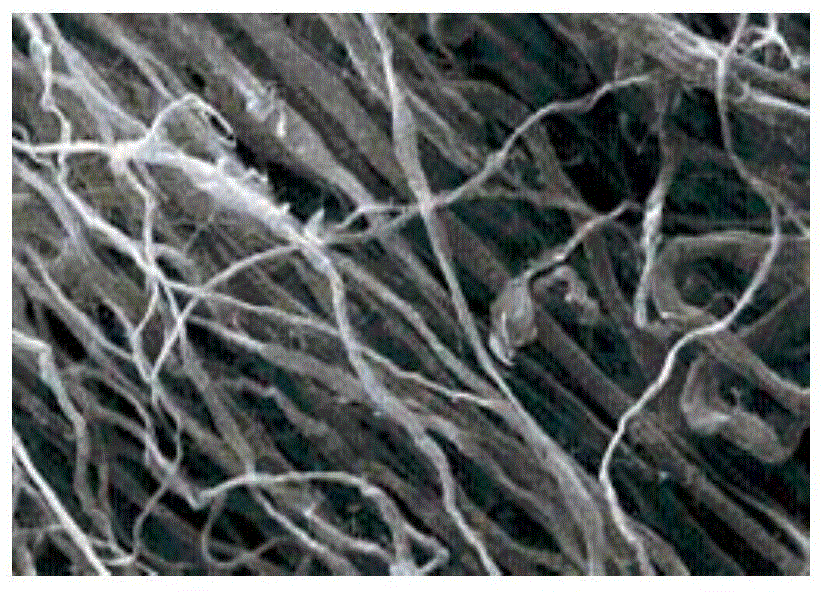
为此,本发明所要解决的技术问题在于现有技术中硬脑/脊膜替代材料植入人体后降解速度过快或过慢均不利于修复硬脑/脊膜缺损的问题,进而提供一种适用于硬脑/脊膜缺损修复降解要求的硬脑/脊膜生物补片材料,并公开其制备方法和应用。为解决上述技术问题,本发明提供了一种硬脑/脊膜生物补片材料的制备方法,包括取动物膜组织为原材料,并顺次置于表面活性剂溶液、碱液、过氧化物溶液、质量浓度为1-5wt%的缩水甘油醚类环氧交联剂溶液以及磷酸盐缓冲液中进行浸泡的步骤,并将经上述各步骤浸泡后的所述膜组织材料冷冻干燥后经辐照灭菌,即得。所述的硬脑/脊膜生物补片材料的制备方法,所述缩水甘油醚类环氧交联剂溶液浸泡的步骤中,所述交联剂为丙三醇缩水甘油醚和/或乙二醇缩水甘油醚。所述的硬脑/脊膜生物补片材料的制备方法,所述缩水甘油醚类环氧交联剂溶液浸泡的步骤中,所述浸泡时间为15h-10d。优选的,所述膜组织材料在浓度为2-4wt%的交联剂中浸泡20h。更优选的,所述膜组织材料在浓度为2wt%的交联剂中浸泡。所述的硬脑/脊膜生物补片材料的制备方法,所述含表面活性剂溶液浸泡的步骤中,所述表面活性剂的质量浓度为0.1-5wt%,并控制所述溶液温度为0-25℃进行浸泡。所述的硬脑/脊膜生物补片材料的制备方法,所述碱液浸泡的步骤中,所述碱液的浓度为0.1-5mol/L,并控制所述溶液温度为5-15℃进行浸泡。优选的,在所述碱液浸泡的步骤中,膜组织材料加入至浓度为1mol/L的氢氧化钠溶液中浸泡2h,并控制溶液温度为8℃。所述的硬脑/脊膜生物补片材料的制备方法,所述过氧化物溶液浸泡的步骤中,所述过氧化物溶液的质量浓度为1-5wt%。优选的,在所述过氧化物溶液浸泡的步骤中,浸泡后的膜组织材料加入至浓度为2wt%过氧化物溶液中浸泡处理1-5h。优选的,所述过氧化物溶液为过氧化氢溶液。所述的硬脑/脊膜生物补片材料的制备方法,所述辐照灭菌的步骤为电子束辐照灭菌,辐照计量为10-30KGy。优选的,所述电子束的辐照计量为25KGy。优选的,在所述表面活性剂溶液浸泡的步骤中,所述表面活性剂为TritonX_100、Tween80或Tween40中的至少一种。优选的,所述动物膜组织原材料为出生6个月以下的猪或牛的膜组织材料。所述磷酸盐缓冲液浸泡的步骤中,所述磷酸盐缓冲液的pH值为5.0-8.0。优选的,在所述磷酸盐缓冲液浸泡的步骤中,所述浸泡后的膜组织加入pH为6.8的磷酸盐缓冲液中浸泡至少5h。本发明提供了一种由上述的方法制备得到的硬脑/脊膜生物补片材料。本发明还提供了一种所述的硬脑/脊膜生物补片材料在制备防止或减少组织粘连材料、用于修复受损的硬脑/脊膜组织的材料、治疗或预防因外伤或肿瘤造成的硬脑/脊膜损伤或缺损的材料、治疗或预防因硬脑/脊膜缺损引起的脑脊液漏、感染材料中的应用。本发明还提供了一种缩水甘油醚类环氧交联剂用于制备硬脑/脊膜生物补片材料的用途。本发明所述的技术方案相比现有技术具有如下优势:(1)本发明所述的硬脑/脊膜生物补片材料制备方法,利用质量浓度为1-5wt%缩水甘油醚类环氧交联剂浸泡处理膜组织材料,获得的生物补片材料特别适用于硬脑/脊膜缺损的修复,在植入人体或动物体内后,能够在体内保持6月-2年时间内稳定不降解,当超过2年后即降解,保证具有一定力学性能以及防液体渗漏作用的同时还具有生物相容性,产生术后并发症以及副作用的风险大大降低,满足了对于用于修复硬脑/脊膜缺损材料的特殊要求;(2)本发明所述的硬脑/脊膜生物补片材料制备方法,通过选择丙三醇缩水甘油醚和/或乙二醇缩水甘油醚中为交联剂,不会增加该硬脑/脊膜生物补片本身的细胞毒性,降低其带来副作用的风险,在使用上述交联剂的补片,细胞毒性虽有略微提升,但是其提升幅度较小,可以忽略不计,并且其毒性都在0级范围内,并不会对被植入的人体或动物产生不利的影响;(3)本发明所述的硬脑/脊膜生物补片材料制备方法,通过碱液以及表面活性剂配合使用获得的生物补片材料脂肪含量明显降低,大大提高了生物补片的安全性和有效性。附图说明为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中图1是未交联的膜材料的扫描电镜照片;图2是交联的膜材料的扫描电镜照片;图3为植入未交联的生物补片的对照组照片;图4为植入实施例1制备的硬脑/脊膜生物补片的试验组的照片;图5本发明实施例1制备的生物补片的HE染色图;图6为未使用硬脑/脊膜修复的对照组;图7为使用实施例1制备的硬脑/脊膜生物补片修复的试验组。具体实施方式实施例1本实施所述的硬脑/脊膜生物补片的制备方法包括如下步骤:(1)取出生6个月以下的牛的膜组织为原材料,进行清洗,去除不适宜加工的部分,备用;(2)取上述清洗后的膜组织加入质量浓度为0.1wt%的TritonX_100溶液中浸泡,震荡过夜,并控制溶液温度为0℃;(3)取上述步骤浸泡后的膜组织加入浓度为5mol/L的氢氧化钠溶液中浸泡2h,并控制溶液温度为15℃;(4)取上述步骤浸泡后的膜组织加入质量浓度为1wt%过氧化氢溶液中浸泡处理1h;(5)取上述步骤浸泡后的膜组织加入浓度为1wt%丙三醇缩水甘油醚中浸泡15h;(6)随后取所述浸泡后的膜组织加入pH为5.0的磷酸盐缓冲液中浸泡至少5h;(7)将上述步骤浸泡后的膜组织冷冻干燥,所述的冷冻干燥程序为:产品预冻先降温至-60℃以下后抽真空、升温,进入升华阶段;一次升华温度由-35℃以下升至-25℃,时间不少于6小时;一次升温温度由-25℃升至-15℃,时间为1-3小时;二次升温温度由-15℃升至25℃,时间为1-3小时。随后以辐照计量为15KGy的电子束辐射灭菌,即得。实施例2本实施所述的硬脑/脊膜生物补片的制备方法包括如下步骤:(1)取出生6个月以下的牛的膜组织为原材料,进行清洗,备用;(2)取上述清洗后的膜组织加入质量浓度为5wt%的Tween80溶液中浸泡,震荡过夜,并控制溶液温度为25℃;(3)取上述步骤浸泡后的膜组织加入浓度为0.1mol/L的氢氧化钾溶液中浸泡4h,并控制溶液温度为5℃;(4)取上述步骤浸泡后的膜组织加入质量浓度为5wt%过氧化物溶液中浸泡处理5h;(5)取上述步骤浸泡后的膜组织加入质量浓度为1wt%乙二醇缩水甘油醚中浸泡15h;(6)随后取所述浸泡后的膜组织加入pH为8.0的磷酸盐缓冲液中浸泡至少5h;(7)将上述步骤浸泡后的膜组织冷冻干燥,所述的冷冻干燥程序为:产品预冻先降温至-60℃以下后抽真空、升温,进入升华阶段;一次升华温度由-35℃以下升至-25℃,时间不少于6小时;一次升温温度由-25℃升至-15℃,时间在1-3小时;二次升温温度由-15℃升至25℃,时间在1-3小时。随后以辐照计量为25KGy的电子束辐射灭菌,即得。实施例3本实施所述的硬脑/脊膜生物补片的制备方法包括如下步骤:(1)取出生6个月以下的牛的膜组织为原材料,进行清洗,备用;(2)取上述清洗后的膜组织加入质量浓度为2wt%的含有TritonX_100和Tween40溶液中浸泡,震荡过夜,并控制溶液温度为8℃;(3)取上述步骤浸泡后的膜组织加入浓度为1mol/L的氢氧化钠溶液中浸泡2h,并控制溶液温度为8℃;(4)取上述步骤浸泡后的膜组织加入质量浓度为2wt%过氧化物溶液中浸泡处理3h;(5)取上述步骤浸泡后的膜组织加入质量浓度为2wt%含有丙三醇缩水甘油醚和乙二醇缩水甘油醚的混合交联剂中浸泡20h;(6)随后取所述浸泡后的膜组织加入pH为6.8的磷酸盐缓冲液中浸泡至少5h;(7)将上述步骤浸泡后的膜组织冷冻干燥,所述的冷冻干燥程序为:产品预冻先降温至-60℃以下后抽真空、升温,进入升华阶段;一次升华温度由-35℃以下升至-25℃,时间不少于6小时;一次升温温度由-25℃升至-15℃,时间在1-3小时;二次升温温度由-15℃升至25℃,时间在1-3小时。随后以辐照计量为30KGy的电子束辐射灭菌,即得。实施例4本实施所述的硬脑/脊膜生物补片的制备方法包括如下步骤:(1)取出生6个月以下的牛的膜组织为原材料,进行清洗,备用;(2)取上述清洗后的膜组织加入质量浓度为3wt%的含有TritonX_100和Tween80溶液中浸泡,震荡过夜,并控制溶液温度为0℃;(3)取上述步骤浸泡后的膜组织加入浓度为1mol/L的氢氧化钠溶液中浸泡4h,并控制溶液温度为8℃;(4)取上述步骤浸泡后的膜组织加入质量浓度为4wt%过氧化物溶液中浸泡处理4h;(5)取上述步骤浸泡后的膜组织加入质量浓度为5wt%含有丙三醇缩水甘油醚和乙二醇缩水甘油醚的混合交联剂中浸泡10d;(6)随后取所述浸泡后的膜组织加入pH为8.0的磷酸盐缓冲液中浸泡至少5h;(7)将上述步骤浸泡后的膜组织冷冻干燥,所述的冷冻干燥程序为:产品预冻先降温至-60℃以下后抽真空、升温,进入升华阶段;一次升华温度由-35℃以下升至-25℃,时间不少于6小时;一次升温温度由-25℃升至-15℃,时间在1-3小时;二次升温温度由-15℃升至25℃,时间在1-3小时。随后以辐照计量为10KGy的电子束辐射灭菌,即得。对照例1本实施例与实施例1的制备方法相同,其区别仅在于没有经过交联剂处理的步骤。对照例2本实施例与实施例2的制备方法相同,其区别仅在于没有经过交联剂处理的步骤。对照例3本实施例与实施例1的制备方法相同,其区别仅在于没有经过所述表面活性剂浸泡的步骤。对照例4本实施例与实施例1的制备方法相同,其区别仅在于所用交联剂为戊二醛交联。效果例以下通过实验例来考察本发明下述相关实施例及对照例制备的硬脑/脊膜生物补片材料各项性能,进而证明本发明制备的硬脑/脊膜生物补片具有显著技术效果。1、撕裂力测试试验方法:使用拉伸(压缩)试验机,将按照实施例1-2、对照例1-2方法制备的生物补片材料,将样品剪成规格为1cm×3cm的长条状,在离短边边缘3-5mm处用4-0号缝合线穿过样品,对折缝合线,在距离穿孔处约5cm处将缝合线打结,防止缝合线脱落;将上述方法得到的试样,用纯化水水化3-5分钟,将未穿线的一端固定在拉力试验机的下部,将穿线一端通过挂钩固定于拉力试验机的上部,开始检测,直至试样被撕裂,读取拉伸负荷力的最大值,即为本试样的撕裂力。结果见下表1以及图1-2。表1撕裂力测试结果测试组撕裂力(N)实施例113.25实施例210.65对照例18.56对照例27.35对照例410.01结论:上表为三次试验共30片产品撕裂力的平均值,实施例1、2无论15KGy还是25KGy的剂量下,相对于无交联剂处理的对照例1、2的情况,撕裂力均有明显提升,提升比例分别为54.79%和44.90%,而相对于采用其他交联剂处理的对照例4,相对于无交联剂处理的对照例1、2的情况,同样撕裂力均有明显提升,提升比例分别为16.94%和36.19%。由于膜组织材料的胶原纤维无规编织而形成的纤维之间存在大量不规则的间隙,孔隙率主要取决天然胶原纤维的结构,交联剂会在胶原间形成新的键将胶原纤维拉紧引起膜材料的轻微收缩,从图1-2中对比可知,交联前后,生物补片的胶原纤维有明显的收缩,证明使用本发明的交联剂有交联效果,对膜的胶原纤维有一定的拉紧作用。2、体外降解方面试验方法:将按照实施例1-2、对照例3-4方法制备的生物补片分剪成(1-3)*(4-6)mm大小的长条状,分别放置于试管中,加入一定量的生理盐水,放置在37℃中1d后,更换一次溶液,然后放置3-4d,取溶液根据医疗器械行业标准16886内的检测方法进行检测,试验结果见下表2。表2降解质量百分比测试组第一次取样第二次取样实施例13.5%1.4%实施例26.2%2.4%对照例115.1%7.3%对照例221.8%7.6%对照例44.1%1.8上面表格表明分别为15KGy和25KGy辐照剂量生物补片的降解质量百分比,通过比较可以看出在有交联剂作用的情况下,生物补片的降解速度明显下降。3、体内降解方面试验方法:将按照实施例1、对照例1、对照例4方法制备的生物补片,将上述制备的生物补片原位植入健康的大鼠体内,所述植入试验方法如下:取20只雄性SD大鼠,3-4周龄,体重180-220g,麻醉后皮下植入实施例1的生物补片,在对侧植入对照例1或对照例4的生物补片,每张材料1cm2,选取植入3个周、12个周、30个周后各5只大鼠处死,取所述植入区的周围组织用多聚甲醛固定照相。试验结果见表3以及图3-4,图3为对照例1的植入12个周后的小鼠照片,图4为实施例1的植入12个周后的小鼠照片,从图中可以看出,图3已完全降解,图4还有未降解的生物补片存在,且膜形态比较完整,膜与周围组织未发生粘连情况,降解时间明显比未交联或选用其他交联剂制备的生物补片延长。试验结果见下表3。表3体内降解试验结果测试组3个月12个月30个月实施例1未降解未完全降解完全降解对照例1未完全降解完全降解——对照例4未降解未完全降解未完全降解4、细胞毒性方面试验方法:将按照实施例1、对照例1、对照例4的方法制备的生物补片进行浸提,制备浸提液;制备相应的细胞悬液;弃去原培养基,在96孔板中加入适当浓度的浸提液、阴性对照、阳性对照以及空白对照,置于37℃二氧化碳培养箱内分别培养24h、48h。在24h和48h后,观察细胞生长状况,弃去96孔培养板内的浸提液,并加入1mg/ml的MTT溶液50μL。加入MTT溶液的96孔板置于37℃二氧化碳培养箱2小时。2小时后,弃掉96孔板中的MTT溶液,并加入异丙醇100μL,震荡96孔板5-10min,测定其在492nm处的吸光度值。细胞毒性的检测方法是参照16886行业标准中细胞毒性检测中的MTT法进行的检测。结果见下表4,表4细胞毒性试验结果测试组观察期24h观察期48h实施例1103.12104.07对照例1100.31101.25对照例492.0290.84上表为三次试验产品细胞毒性的平均值,可以清楚的看到实施例1使用交联剂制备的产品,细胞毒性有所降低,且都在0级范围内,相比于对照例1的细胞毒性实施例1的下降比例为2.80%和2.79%,细胞毒性变化不大,说明加入此交联剂后,不会增加产品本身的细胞毒性,然而相比于对照例1的细胞毒性对照例4的细胞毒性有一定的增加,且增加比例为9.01%和11.46%,说明对照例4中所用交联剂有一定的细胞毒性。5、脂肪含量方面试验方法:将按照实施例1、对照例3方法制备的生物补片进行试验,脂肪含量的测定方法是依据《中华人民共和国药典》二部“胰酶脂肪检测”制定,测定结果见下表5。表5脂肪含量试验结果对照例3实施例1提升比例脂肪含量0.27%0.06%77.77%上表为三次试验0.1g生物补片脂肪含量的平均值,可以清楚的看到本发明实施例1通过碱液NaOH和表面活性性剂TrironX-100配合使用获得的生物补片,脂肪含量明显降低。6、安全性和有效性6.1安全性试验方法:将本发明实施例1制备的生物补片进行HE染色处理,所述HE染色处理步骤如下:将材料用梯度酒精下行入水,后用自来水和蒸馏水先后清洗,用0.5%苏木素处理后用自来水清洗,再用1%盐酸酒精分色后用自来水清洗,再用1/400的氨水促蓝后清洗,用1%伊红染色后用80%酒精分色,梯度酒精脱水,二甲苯透明后得到树脂封片。本发明实施例1制备的生物补片材料的HE染色图见图5,从图中可知,该生物补片没有细胞和细胞核残留,证明本发明的方法可以有效的脱去膜材料中的细胞等杂物,安全性高。6.2有效性试验方法:将本发明实施例1制备的生物补片植入健康白兔中作为试验组,以未使用硬脑脊膜修复的兔子作为对照组,所述试验方法如下:麻醉兔子后,打开兔子头盖骨,破坏硬脑膜,加入生物补片替代硬脑膜,对照组不植入生物补片;然后将头盖骨复位,缝合伤口。定期查看实验结果,试验结果见图6-7,图6为未使用硬脑脊膜修复的对照组,图7为使用本发明实施例1制备的硬脑/脊膜生物补片的试验组照片,从图中可以看出两张图片的伤口都已经痊愈,但图6照片中疤痕较大,且有部分组织出现坏死现象,而图7照片中伤口恢复良好,且疤痕较小,伤口平滑,没有组织坏死现象,说明修复组织功能良好且无免疫排斥反应。显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1