聚氨酯/小肠粘膜下层复合材料的制备方法与流程
本发明涉及聚氨酯/小肠粘膜下层复合材料的制备方法。
背景技术:
理想的软组织修复材料应该具有下述性质:(1)生物相容性,例如细胞相容性、组织相容性等;(2)优良的力学性能,例如拉伸强度、回弹性等。此外,软组织修复材料还需要适宜的三维多孔结构,从而有利于细胞的长入和分化、组织再生以及营养物质的传送。目前,软组织修复材料主要有两大类:(1)合成高分子材料,例如聚氨酯等,具有优良的物理机械性能,但是不具备生物活性,不能促进组织再生;(2)天然的细胞外基质(extracellularmatrix,ECM),例如胶原、明胶、小肠粘膜下层(smallintestinalsubmucosa,SIS)等,主要由胶原蛋白、纤维蛋白、弹性蛋白、生长因子、氨基葡聚糖或信号分子等多种成分组成,具有优良的生物相容性,易降解,降解产物无毒副作用、炎性反应低,可诱导和促进组织结构的再生和修复,但是存在回弹性差、易塌陷的问题。因此,亟需发明一种新的材料,能够同时具有优良的物理机械性能和生物活性,从而克服现有软组织缺损修复材料只具有单一方面的性能以及应用受到限制等缺陷。
技术实现要素:
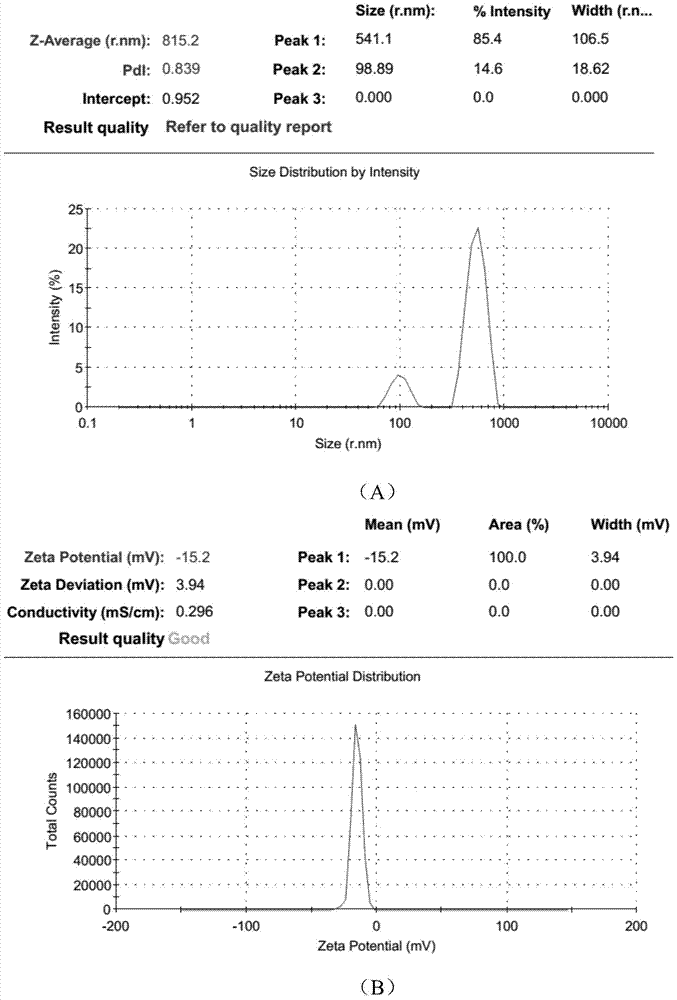
本发明的目的在于提供聚氨酯/小肠粘膜下层(PU/SIS)复合材料的制备方法,制备得到的复合材料同时具有聚氨酯和SIS的优良性能,既具有良好的回弹性能、力学性能,又具有生物活性、生物相容性,可诱导和促进组织结构的再生和修复,克服了现有软组织缺损修复材料只具有单一方面的性能以及应用受到限制等缺陷。本发明是通过下述技术方案实现的:聚氨酯/小肠粘膜下层复合材料的制备方法,包括由以下步骤组成:a、将阴离子水性聚氨酯乳液与小肠粘膜下层混匀,真空冻干成型;其中,小肠粘膜下层的用量是聚氨酯乳液的0.01~0.03倍(w/w),所述聚氨酯乳液的平均粒径为15.18nm~42.86nm,固含量为3%~21%;b、交联,真空冻干,即得。所述的制备方法,步骤a中,所述聚氨酯乳液由如下方法制备得到:(1)预聚:取异氰酸酯与羟基供体,预聚,二者的摩尔比为(1.45~1.85):0.5;优选地,异氰酸酯与羟基供体的摩尔比为1.45:0.5;所述异氰酸酯为异佛尔酮二异氰酸酯、L-赖氨酸二异氰酸酯、二苯基甲烷二异氰酸酯中的任意一种或多种;优选地,所述异氰酸酯为异佛尔酮二异氰酸酯;所述羟基供体为聚乙二醇、聚四氢呋喃、丙二醇、1,4-丁二醇中的任意一种或多种;优选地,所述羟基供体为聚四氢呋喃;(2)扩链:加入扩链剂,扩链,其中,扩链剂与步骤(1)中羟基供体的摩尔比为(3~7):3;所述扩链剂为2,2-二羟甲基丁酸、2,2-二羟甲基丙酸、N-甲基二乙醇胺、二乙醇胺中的任意一种或多种;优选地,所述扩链剂为2,2-二羟甲基丁酸或2,2-二羟甲基丙酸;(3)中和乳化:加入中和剂,搅匀,然后将反应体系滴加到20%(v/v)的丙酮水溶液中,高速搅拌1~2h;其中,中和剂与步骤(2)中扩链剂的摩尔比为1.5:1;高速搅拌的转速为1200~1400rpm;所述中和剂为三乙胺或氢氧化钠;(4)纯化:旋蒸除去丙酮和过量中和剂,去离子水透析除去杂质,即得。步骤(1)中,预聚的温度为70℃~80℃;预聚的时间为1~5h;预聚采用的催化剂为辛酸亚锡、三乙醇胺或二月桂酸二丁基锡;步骤(2)中,扩链的温度为50℃~55℃;扩链的时间为1~5h;步骤(3)中,高速搅拌的转速为1300rpm;高速搅拌的时间为2h;步骤(4)中,旋蒸的温度为55℃;优选地,步骤(1)中,预聚的温度为74℃,预聚的时间为2h50min;步骤(2)中,扩链的温度为54℃,扩链的时间为3h。所述的制备方法,步骤a中,小肠粘膜下层的用量是聚氨酯乳液的0.03倍(w/w)。所述的制备方法,步骤a中,所述小肠粘膜下层按照下述方法制备得到:(i)取猪小肠,水洗,剖开,剪成段;(ii)刮除小肠的肌层和浆膜层;(iii)水洗滤干后,将其浸入三氯甲烷与甲醇的混合溶液中,浸泡时间为4小时,所述混合溶液中三氯甲烷与甲醇的体积比为1:1;(iv)水洗后,放入胰蛋白酶溶液中,4℃处理过夜;(v)水洗后,用SDS处理至少4h;(vi)清洗后,冻干;(vii)粉碎,加入含有3%醋酸、0.1%胃蛋白酶的PBS溶液中,调pH至中性后,冻干,再粉碎,即得。所述的制备方法,步骤a和b中,真空冻干的温度为-40~-80℃。所述的制备方法,步骤b中,交联时间为12~48h。所述的制备方法,步骤b中,所述交联的方法是:将步骤a制得的材料浸泡于交联剂溶液中;其中,交联剂是1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、京尼平或戊二醛;优选地,所述交联剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐或京尼平;所述交联剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐时,交联剂溶液的浓度为1%~5%,优选为2.5%,pH值为3~9,优选为5.5~7.4;所述交联剂为京尼平时,交联剂溶液的浓度为0.1%~2%,优选为2%,pH值为3~9,优选为7.4。本发明PU/SIS复合材料的制备方法,具有下述有益效果:(1)本发明方法制备的PU/SIS复合材料,具有优良的力学性能,其拉伸强度和断裂伸长率均较高,可以满足软组织缺损修复材料对力学性能的要求;(2)本发明方法制备的PU/SIS复合材料,回弹性好,力学性能更加均一可控,克服了现有技术中小肠粘膜下层应用于软组织修复材料存在的回弹性差、易塌陷等缺陷;(3)本发明方法制备的PU/SIS复合材料,为三维多孔结构,孔径均匀,且孔洞间相互贯通,孔径大小在30-150μm之间,平均孔径为62.44±23.74μm,适合细胞的粘附爬行,有利于诱导细胞和毛细血管的长入,促进细胞的增殖;(4)本发明方法制备的PU/SIS复合材料,组织相容性好,免疫原性低;(5)本发明PU/SIS复合材料的制备方法,还具有操作简便、成本低廉等优点。综上,本发明方法制备的PU/SIS复合材料,同时具有聚氨酯和小肠粘膜下层的优良性能,既具有良好的回弹性能、力学性能,又具有生物活性、生物相容性,可诱导和促进组织结构的再生和修复,克服了现有软组织缺损修复材料只具有单一方面的性能以及应用受到限制等缺陷;同时,本发明方法制备的PU/SIS复合材料,为三维多孔结构,孔径均匀,且孔洞间相互贯通,生物相容性好,免疫原性低,具有良好的应用前景。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1实施例1与4聚氨酯乳液平均粒径及Zeta电位的检测结果。图2实施例1~4中SIS水溶液平均粒径及Zeta电位的检测结果。图3实施例2与3聚氨酯乳液平均粒径及Zeta电位的检测结果。图4聚氨酯乳液的红外光谱图:A:实施例1与4聚氨酯乳液的红外测试结果;B:实施例2与3聚氨酯乳液的红外测试结果。图5试验例1对比试验的结果附图。图6实施例1~4中PU/SIS复合材料的拉伸强度和断裂伸长率的测试结果A:拉伸强度;B:应力应变曲线。图7PU/SIS复合材料的压缩强度、压缩模量以及压缩循环曲线的测试结果A:PU与PU/SIS弹性体生物材料的压缩强度;B:PU与PU/SIS弹性体生物材料的弹性模量;C:PU的压缩循环曲线(n=1);D:PU/SIS弹性体生物材料的压缩循环曲线(n=1);E:PU的压缩循环曲线(n=3);F:PU/SIS弹性体生物材料的压缩循环曲线(n=3)。图8PU/SIS复合材料的扫描电镜检测结果:A:×100;B:×500;C:平均孔径分布图。图9HUVECs在PU/SIS复合材料、PU材料及对照组的生长增殖情况:A:材料对HUVECs细胞吸光值的影响;B:材料对HUVECs细胞存活率的影响。图10HUVECs种植于PU及PU/SIS复合材料上活死细胞染色;A:PU材料培养第3天(×200),B:PU材料培养第5天(×200),C:PU材料培养第8天(×200)D:PU/SIS复合材料培养第3天(×200),E:PU/SIS复合材料培养第5天(×200),F:PU/SIS复合材料培养第8天(×200)。图11SD大鼠背部皮下植入PU及PU/SIS复合材料HE染色结果:A~C为PU材料在第2,4,8周时炎性细胞情况(×200);D~F为PU/SIS复合材料在第2,4,8周时炎性细胞情况(×200)。图12SD大鼠背部皮下植入PU及PU/SIS复合材料Masson染色结果:A-C为PU材料在第2,4,8周的纤维囊厚度情况(×200);D-F为PU/SIS复合材料在第2,4,8周时纤维囊厚度情况(×200)。具体实施方式本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。缩略词:IPDI:异佛尔酮二异氰酸酯,PTMG1000:聚四氢呋喃1000,DMBA:2,2-二羟甲基丁酸,DMPA:2,2-二羟甲基丙酸,TEA:三乙胺,SIS:小肠粘膜下层,PU:聚氨酯,EDC:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,HUVECs:人脐静脉内皮细胞。1.聚氨酯乳液固含量的测定固含量是乳液或涂料在规定条件下烘干后剩余部分占总量的百分数。参照GB1725-79,取干燥的表面皿称质量,并用减重法称取1.5~2.0g的水性聚氨酯乳液平铺于表面皿中,放入40℃烘箱中干燥24h,取出、冷却至室温后称量。用分析天平精确称量前洗净干燥的称量瓶称重Ag,样品重量Bg,烘干后称重(包含了样品和称量瓶)Cg,计算(C-A)/B*100%得出固含量。2.粒径表征与Zeta电位表征2.1平均粒径检测方法采用粒径测试仪对液体样品的平均粒径大小及分布进行测试。取适量制备的水性聚氨酯乳液或1%的SIS水溶液,用移液枪取1ml样品转移至样品池中。将装有液体样品的样品池放入仪器光路中,在室温下进行平均粒径测试。2.2Zeta电位检测方法:采用Zeta电位分析仪对液体样品的Zeta电位进行测试。取适量制备的水性聚氨酯乳液或浓度为1%的SIS水溶液,用移液枪取1ml左右样品转移至样品池中。将装有液体样品的样品池放入仪器光路中,在室温下进行Zeta电位测试。3傅里叶红外光谱表征采用傅里叶红外光谱仪对制得的聚氨酯乳液冷冻干燥后进行傅里叶红外光谱分析。扫描范围400cm-1-4500cm-1,测试方法:ATR模式。实施例1本发明制备PU/SIS复合材料的方法1、制备阴离子水性聚氨酯乳液1)预聚:将摩尔比为1.45:0.5的异佛尔酮二异氰酸酯(IPDI)和聚四氢呋喃1000(PTMG1000),其中异佛尔酮二异氰酸酯为0.1mol,加入三颈瓶中,加入0.2ml辛酸亚锡催化剂,在74℃下预预聚2h50min;2)扩链:将PTMG1000/DMBA摩尔比为5:5的2,2-二羟甲基丁酸(DMBA)在54℃下反应3h。反应过程中观察体系粘度,必要时可加入适量丙酮调节反应体系的粘度,目测反应体系在搅拌过程中不在搅拌杆上聚集的粘度均可,使反应维持在平稳状态下进行;3)中和乳化:室温下,将TEA/DMBA摩尔比为1.5:1的三乙胺加入到体系中,搅拌20min后滴加到250ml丙酮水溶液(丙酮:去离子水=1:4)中,在转速为1300rpm的条件下高速搅拌2h;4)纯化:55℃温度下旋蒸,除去乳液中的丙酮和三乙胺,去离子水透析三天除去杂质。对聚氨酯乳液分别进行平均粒径检测和Zeta电位检测以及傅里叶红外光谱分析,结果分别如图1(A)、(B)以及图4(A)所示。由图1可知,聚氨酯乳液的平均粒径为42.86nm,Zeta电位为-40.4mV。由图4A可知,3329cm-1处为氨基甲酸酯结构中N-H的伸缩振动吸收峰;2750-3000cm-1处为-CH2和-CH3的伸缩振动峰;1712cm-1附近处的吸收峰为氨酯基中的C=O的伸缩振动峰;1112cm-1附近处为C-O-C的伸缩振动峰,2240-2270cm-1区域内异氰酸根基团特征吸收峰基本消失,说明聚氨酯体系中的异氰酸根已经完全参与了反应,这些特征峰的出现或消失表明实验成功合成了聚氨酯。制备得到的聚氨酯乳液的固含量一般较高,可以用蒸馏水稀释成所需要的浓度。2、SIS粉末的制备本发明中,SIS粉末采用现有技术的常规方法制备得到,例如采用下述方法制备SIS粉末:1)取新鲜猪小肠,用水冲去内容物,盐揉搓后用水反复冲洗3次。用手术刀剖开小肠,并剪成10~20cm长的肠段;2)用压舌板刮除小肠的肌层和浆膜层,4℃下保存于生理盐水中;3)去离子水漂洗干净并滤干后将其浸入三氯甲烷-甲醇的混合液中(三氯甲烷:甲醇=1:1)4小时,平均2h换一次液,0.5h搅拌一次;4)去离子水反复清洗后放入浓度为0.25%的胰蛋白酶液里,4℃处理过夜;5)用去离子水漂洗10次后用0.5%的SDS处理至少4h;6)清洗后置于–70℃下冻干24h。7)将低温粉碎的SIS加入含有3...
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1