引物组、锚定引物、试剂盒、文库构建及基因测序方法与流程
本发明涉及分子生物学领域,更具体地说,涉及一种引物组、锚定引物、文库构建方法及基因测序方法。
背景技术:
第二代高通量测序技术包括连接测序法和合成测序法。其中,所述连接测序法是基于连接酶在核酸片段之间进行连接反应的过程中的保真性来实现的,以待测序核酸片段为模板,锚定引物(又称测序引物,其与待测序核酸片段所在链互补)和寡核苷酸探针(该探针的特定位置上带有荧光标记)进行连接反应,通过检测连接产物上的荧光标记从而确定寡核苷酸探针上带有荧光标记的特定位置对应的序列的信息。所述合成测序法是基于聚合酶在延伸核酸链过程中的保真性来实现的,以待测序核酸片段为模板,锚定引物互补结合至待测序核酸片段上,通过检测在延伸过程中产生的信号来确定待测序核酸片段上相应位置的序列信息。第二代高通量测序技术因为其高通量和低成本,目前用于snp分型检测。
现有技术中的一种利用第二代高通量测序技术的snp分型方法,包括以下步骤:a、通过引物扩增获得待测snp位点的核酸序列;b、基于步骤a的产物构建测序文库;c、将锚定引物锚定在测序文库分子上,通过高通量测序技术检测待测snp位点的序列信息。为了加快待测snp位点的检测,减少测序的循环数,提高snp位点检测效率,一般将锚定引物设计在待测snp位点的附近。但是这种设计方法,往往会因为snp位点所在序列的特殊性,例如该snp位点附近序列存在特殊结构——重复序列、发夹情况等,导致按照上述方法设计的锚定引物,无法准确的锚定在预定位置,造成测序结果不准确或测序失败;使得该方法无法大规模推广。
因此需要一种新的适用范围广的引物组、锚定引物、试剂盒、文库构建及基因测序方法。
技术实现要素:
本发明的目的在于提供一种新的适用范围广的引物组、锚定引物、试剂盒、文库构建及基因测序方法,旨在解决现有的高通量基因测序技术中锚定引物的非特异性锚定的技术问题。
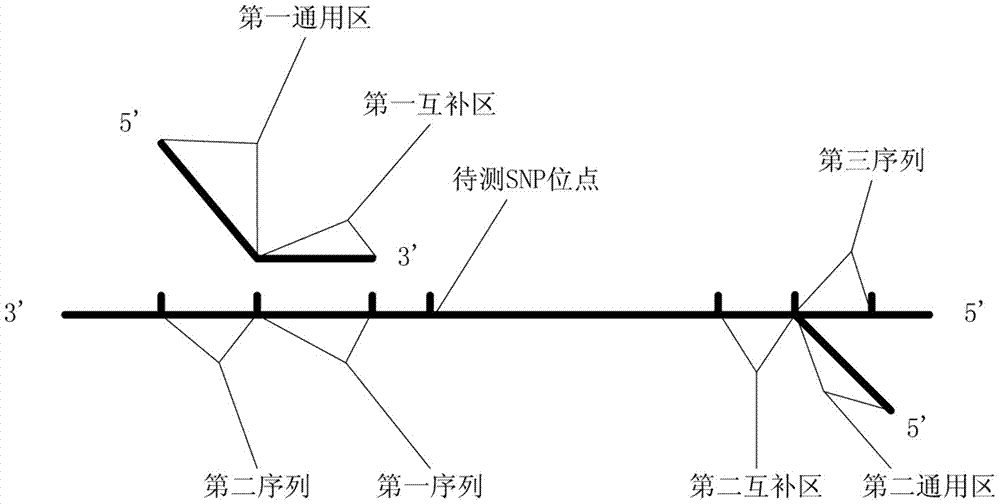
为了实现发明目的,本发明提供了一种引物组,包括第一引物对,所述第一引物对中的上游引物由第一通用区和第一互补区组成;所述第一互补区与所述第一通用区的3’末端连接,所述第一互补区与第一序列完全互补;所述第一序列为待测snp位点所在序列中的一段序列,处于待测snp位点的3’端,所述第一序列的5’末端距待测snp位点1至7bp;所述第一通用区不与第二序列互补;所述第二序列为待测snp位点所在序列上与第一序列的3’末端连接的一段序列。
优选的,所述第一互补区序列长度在6至12bp之间。
优选的,所述第一通用区序列长度在6至16bp之间。
优选的,所述第一序列的5’末端距待测snp位点1至3bp。
优选的,所述引物组还包括第二引物对,所述第一引物对和第二引物对分别为对待测snp位点所在序列进行扩增的内引物对和外引物对。
优选的,所述第一引物对中的下游引物由第二通用区和第二互补区组成;所述第二互补区与所述第二通用区的3’末端连接;所述第二互补区为待测snp位点所在序列中的一段序列,处于待测snp位点的5’端;所述第二通用区与第三序列不同;所述第三序列为待测snp位点所在序列上与第二互补区的5’末端连接的一段序列。
优选的,所述第一通用区中含有u。
为了更好的实现本发明的目的,本发明还提供了一种锚定引物,所述锚定引物与第四序列完全互补配对;所述第四序列为单链核酸分子,是上述任一种引物组中的第一引物对的扩增产物中的一段序列;所述第四序列包括第一序列和第一归一化区,所述第一归一化区与第一序列的3’末端连接,所述第四序列的5’末端距待测snp位点1至7bp;或所述第四序列包括第一互补区和第二归一化区,所述第二归一化区与第一互补区的5’末端连接,所述第四序列的3’末端距待测snp位点1至7bp。
优选的,所述第四序列包括第一序列和第一归一化区,所述第一归一化区与第一序列的3’末端连接,所述第一序列的5’末端距待测snp位点1至3bp;或
所述第四序列包括第一互补区和第二归一化区,所述第二归一化区与第一互补区的5’末端连接,所述第四序列的3’末端距待测snp位点1至3bp。
优选的,所述第一互补区序列长度在6至12bp之间。
优选的,所述第一归一化区的长度在8至14bp之间。
为了更好的实现本发明的目的,本发明还提供了一种试剂盒,包括上述的任一种引物组。
优选的,所述试剂盒还包括上述的任一种锚定引物。
为了更好的实现本发明的目的,本发明还提供了另一种试剂盒,包括上述的任一种锚定引物。
优选的,所述试剂盒还包括上述的任一种引物组。
为了更好的实现本发明的目的,本发明还提供了一种文库构建方法,包括以下步骤:
a、利用上述的任一种引物组,对待测样本进行pcr扩增,得含待测snp位点的扩增产物;
b、将含待测snp位点的扩增产物与接头连接,形成待测序文库分子。
优选的,所述步骤b为:将含待测snp位点的扩增产物直接与接头连接,形成待测序文库分子,并通过微球可寻址的固定在固相载体上。
更优选的,所述步骤b包括以下步骤:
b1、将含待测snp位点的扩增产物直接与固定在微球上的接头连接,形成固定在微球上的待测序文库分子;
b2、将步骤b1所得微球可寻址的固定在固相载体上。
更优选的,所述步骤b1中的连接反应是在切割-连接反应体系中进行的,所述切割-连接反应体系包括:连接酶、断裂剂、固定在微球上的第一接头和连接缓冲液;所述第一通用区中含有u;所述断裂剂用于特异性切割u,并使含待测snp位点的扩增产物形成第一粘性末端;所述第一接头为核酸分子,含有与第一粘性末端完全互补配对的第二粘性末端。
更优选的,步骤b1中所述连接缓冲液中含有peg。
为了更好的实现本发明的目的,本发明还提供了一种基因测序方法,包括以下步骤:
a、利用上述的任一种引物组,对待测样本进行pcr扩增,得含待测snp位点的扩增产物;
b、将含待测snp位点的扩增产物与接头连接,形成待测序文库分子;
c、利用上述的任一种锚定引物,对待测序文库分子进行测序,获得待测snp位点的序列信息。
优选的,所述步骤c中的测序方法为连接测序法;所述步骤b、c之间还包括步骤d,采用上述任一种锚定引物,利用无荧光标记的寡核苷酸探针替换有荧光标记的寡核苷酸探针,对待测序文库分子进行一次连接测序反应。
优选的,所述步骤d包括以下步骤:
d1、将所述锚定引物锚定在待测序文库分子上;
d2、加入无荧光标记的寡核苷酸探针、连接酶以及相应的缓冲液,进行连接反应;
d3、变性去除连接产物。
由上可知,本发明通过对待测snp位点的扩增引物进行特殊设计,使得通过该扩增引物扩增的产物中的待测snp位点附近均包括第一通用区,降低不同待测snp位点附近区域的复杂度,然后基于第一通用区与待测snp位点之间的序列,可设计相应的锚定引物,使锚定引物准确的锚定在目标位置,本发明的引物组、锚定引物、试剂盒、文库构建及基因测序方法适用于多种不同的snp位点的检测,尤其适用于多snp位点的同时检测,能在保证snp位点检测效率的前提下,避免因为待测snp位点附近序列的特殊性而导致的测序结果不准确或测序失败现象的出现。
附图说明
图1是本发明第一典型实施例中第一引物对中的上游引物与待测snp位点所在序列之间的关系示意图。
图2是本发明一实施例中第一引物对中的下游引物与待测snp位点所在序列之间的关系示意图。
图3是本发明第二典型实施例中的锚定引物与第一引物对扩增产物之间的关系示意图。
图4是本发明第一具体实施例中第一接头的结构示意图。
图5是本发明第一具体实施例中第一接头的标签序列与样本来源和snp位点的对应关系图。
图6是本发明第一具体实施例中实验组测序流程图。
图7是本发明第一具体实施例中第一对比实验的测序流程图。
图8是本发明第一具体实施例中样本2的初步实验结果对照图。
图9是本发明第三对比实验中rs671和rs1801253位点的测序采图结果。
图10是本发明第一具体实施例中rs671和rs1801253位点的测序采图结果。
图11是本发明第三对比实验中rs1799853位点的测序采图结果。
图12是本发明第一具体实施例中rs1799853位点的测序采图结果。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。
本发明提出第一典型实施例,一种引物组,包括第一引物对,如图1所示,所述第一引物对中的上游引物由第一通用区和第一互补区组成;所述第一互补区与所述第一通用区的3’末端连接,所述第一互补区与第一序列完全互补;所述第一序列为待测snp位点所在序列中的一段序列,处于待测snp位点的3’端,所述第一序列的5’末端距待测snp位点1至7bp;所述第一通用区不与第二序列互补;所述第二序列为待测snp位点所在序列上与第一序列的3’末端连接的一段序列。
需要说明的是,所述引物组可用于对待测snp位点所在序列进行扩增,所得扩增产物可用于构建含待测snp位点的测序文库,进而进行待测snp位点的高通量测序检测。本发明通过对第一引物对中的上游引物的特殊设计,使得基于第一引物对的扩增产物中,待测snp位点附近增加了一个含第一通用区的序列。所述第一通用区的序列既不是待测样本的模板分子中待测snp位点附近的序列,也不是待测样本的模板分子中待测snp位点附近的互补序列。所述第一通用区使得针对待测snp位点的扩增产物的一端增加了一个归一化序列。所述第一通用区不与第二序列互补,无重复序列,自身不会形成发夹。本方案尤其适用于待测样本中的模板分子上的待测snp位点附近序列结构复杂的待测样本,能够有效简化待测snp位点附近的序列结构,降低锚定引物的设计难度,可有效保证锚定引物准确的锚定在目标位置,避免因为待测snp位点附近序列的特殊性而导致的测序结果不准确或测序失败现象的出现,适用于各类型待测snp位点的检测。针对不同的待测snp位点,该第一通用区序列可相同。
另外,第一序列的5’末端距待测snp位点1至7bp,使得基于所述第一引物对扩增产物所得待测序文库分子进行高通量基因测序时,锚定引物的末端与待测snp位点之间的距离在1至7bp之间,当利用连接测序法和t4连接酶进行测序时,可保证仅通过一次锚定引物与特定位置含荧光标记的探针的连接,即可实现对待测snp位点的检测,提高高通量基因测序的效率。
在本发明的一个实施例中,所述第一序列的5’末端距待测snp位点1至3bp。
本方案可有效控制待测snp位点与第一通用序列之间的间距,使得锚定引物中含有一段足够长度的第一通用序列或第一通用序列的互补序列;这在同时检测多个待测snp位点时,既保证各锚定引物均能准确锚定在各自目标位置上,又使得各锚定引物与目标位置结合情况更为一致,归一化,减小因为各锚定引物与目标位置结合情况的差异,从而减小各待测snp位点检测信号的差异,提高测序的准确性。
为了保证利用第一引物对进行扩增所得产物的纯度,避免非特异性扩增,本发明在第一典型实施例的基础上,提出一实施例,所述第一互补区序列长度在6至12bp之间。
本方案保证了第一引物对中的上游引物与含待测snp位点的模板序列的特异性集合,避免了非特异性扩增,又保证了不同待测snp位点附近序列之间具有足够的区分度。更优选的,所述第一互补区序列长度在7至10bp之间。
为了进一步保证降低了不同待测snp位点附近区域的序列复杂度,本发明在上述实施例的基础上提出另一实施例,所述第一通用区序列长度在6至16bp之间。
本方案中,第一通用序列较大程度的避免了因为序列过长或序列过短导致的第一通用序列内部形成二级结构,或第一通用序列与其他序列之间形成二级结构的可能性,降低了锚定引物的设计难度。
在上述任一实施例的基础上,本发明提出另一实施例,所述引物组还包括第二引物对,所述第一引物对和第二引物对分别为对待测snp位点所在序列进行扩增的内引物对和外引物对。
本方案中,针对待测snp位点,可利用内引物对和外引物对对待测snp位点所在序列进行巢式pcr扩增,从而提高第一引物对扩增产物中目标分子的纯度。
为了进一步降低利用上述引物组扩增所得产物之间的结构差异,基于上述任一实施例,本发明提出另一具体实施例,如图2所示,所述第一引物对中的下游引物由第二通用区和第二互补区组成;所述第二互补区与所述第二通用区的3’末端连接;所述第二互补区为待测snp位点所在序列中的一段序列,处于待测snp位点的5’端;所述第二通用区与第三序列不同;所述第三序列为待测snp位点所在序列上与第二互补区的5’末端连接的一段序列。
需要说明的是,所述第二通用区的序列既不是待测样本的模板分子中待测snp位点附近的序列,也不是待测样本的模板分子中待测snp位点附近的互补序列。所述第二通用区中无重复序列,自身不会形成发夹。基于本方案的第一引物对扩增所得产物,在待测snp位点两端各有一通用序列,针对不同的待测snp位点,该第一通用区和第二通用区序列可均相同;从而降低了不同待测snp位点附近区域的序列复杂度。同时也使得针对各待测snp位点的扩增效率更为一致。
基于上述任一实施例,为了使得基于第一引物对获得的扩增产物能够更快的完成测序文库的构建,本发明提出一实施例,所述第一通用区中含有可断裂位点。所述可断裂位点能被切割剂特异性切割,使得基于第一引物对获得的扩增产物形成第一粘性末端。
本方案中,基于第一通用区中的可断裂位点,扩增产物被切出第一粘性末端后,可与含有与第一粘性末端互补配对的第二粘性末端的接头分子直接连接,提高连接效率。
优选的,所述可断裂位点为核糖核苷酸、rna序列或限制性内切酶酶切位点。相应的,所述断裂剂为rnaseh、rnaseh或限制性内切酶。
当所述可断裂位点为u时,所述断裂剂优选为user酶。
优选的,所述可断裂位点与其所在链5’末端的距离为1至6个碱基。此时,第一粘性末端与接头的第二粘性末端的互补配对连接反应效率较高,更优选为4或5bp,此时连接反应的效率最高。
为了更好的实现本发明的目的,本发明提出第二典型实施例,如图3所示,一种锚定引物,所述锚定引物与第四序列完全互补配对;所述第四序列为单链核酸分子,是上述任一种引物组中的第一引物对的扩增产物中的一段序列;如图3a所示,所述第四序列包括第一序列和第一归一化区,所述第一归一化区与第一序列的3’末端连接,所述第四序列的5’末端距待测snp位点1至7bp;或如图3b所示,所述第四序列包括第一互补区和第二归一化区,所述第二归一化区与第一互补区的5’末端连接,所述第四序列的3’末端距待测snp位点1至7bp。
需要说明的是,所述第一归一化区为第一通用区的互补序列中与第一序列的3’末端连接一段序列;所述第二归一化区为第一通用区中与第一互补区的5’端连接的一段序列。本方案中的锚定引物基于上述任一方案中引物组的第一引物对的扩增产物构建的测序文库而设计,针对不同的待测snp位点,这些锚定引物含有一个共同第一归一化序列或第二归一化序列,它们能准确锚定在目标位置上,避免因为待测snp位点附近序列的特殊性而导致的测序结果不准确或测序失败现象的出现,又使得各锚定引物与目标位置结合情况更为一致、归一化,减小因为各锚定引物与目标位置结合情况的差异,从而减小各待测snp位点检测信号的差异,提高测序的准确性。
基于上述实施例,本发明提出另一实施例,所述第四序列包括第一序列和第一归一化区,所述第一归一化区与第一序列的3’末端连接,所述第一序列的5’末端距待测snp位点1至3bp;或所述第四序列包括第一互补区和第二归一化区,所述第二归一化区与第一互补区的5’末端连接,所述第四序列的3’末端距待测snp位点1至3bp。
本方案中,锚定引物上用于延伸的一端与待测snp位点的距离为1至3bp,当使用连接测序技术进行测序时,可保证仅通过一次锚定引物与特定位置含荧光标记的探针的连接,即实现对待测snp位点的检测,提高高通量基因测序的效率,且1至3bp在t4连接酶的保真度范围(1至7bp)中处于高保真的范围,可有效提高待测snp位点的检测准确度;当使用合成测序技术进行测序时,所述第四序列包括第一互补区和第二归一化区,所述第二归一化区与第一互补区的5’末端连接,所述第四序列的3’末端距待测snp位点1bp,此时仅需要延伸1个bp,即进行1个循环的反应,即可实现对待测snp位点的检测,检测效率高。
在本发明的一个实施例中,所述第一互补区序列长度在6至12bp之间。本方案进一步保证了锚定引物与待测snp位点附近的目标位置结合的特异性。更优选的,所述第一互补区序列长度在7至10bp之间。
在本发明的一个实施例中,所述第一归一化区的长度在8至14bp之间。本方案保证了针对不同待测snp位点设计的锚定引物与目标位置的结合效率的一致性。
为了更好的实现本发明的目的,本发明提出第三典型实施例,一种试剂盒,包括上述的任一种引物组。
本方案中的试剂盒可为扩增试剂盒、文库构建试剂盒或测序试剂盒。当所述试剂盒为扩增试剂盒时,其还可包括pcr扩增所需的其他试剂;当所述试剂盒为文库构建试剂盒时,其还可包括文库构建过程中所需的其他试剂,也可包括pcr扩增所需的试剂;当所述试剂盒为测序试剂盒时,其可包括测序过程中所需的其他试剂,也可包括pcr扩增所需的试剂,也可包括文库构建过程中所需的试剂。
在本发明的一个实施例中,所述试剂盒还包括上述的任一种锚定引物。
为了更好的实现本发明的目的,本发明提出第四典型实施例,一种试剂盒,包括上述的任一种锚定引物。
本方案中的试剂盒一般为测序试剂盒,其可包括测序过程中所需的其他试剂,也可包括pcr扩增所需的试剂,也可包括文库构建过程中所需的试剂。
在本发明的一个实施例中,所述试剂盒还包括上述的任一种引物组。
为了更好的实现本发明的目的,本发明提出第五典型实施例,一种文库构建方法,包括以下步骤:
a、利用上述的任一种引物组,对待测样本进行pcr扩增,得含待测snp位点的扩增产物;
b、将含待测snp位点的扩增产物与接头连接,形成待测序文库分子。
需要说明的是,本方案中的待测序文库分子是基于上述的任一种引物组对待测样本进行pcr扩增后所得的扩增产物获得的,因此,在待测snp位点附近增加了一个含第一通用区的序列。所述第一通用区不与第二序列互补,无重复序列,自身不会形成发夹。因此能够有效简化待测snp位点附近的序列结构,降低锚定引物的设计难度,尤其适用于待测样本中的模板分子上的待测snp位点附近序列结构复杂的待测样本,避免因为待测snp位点附近序列的特殊性而导致的测序结果不准确或测序失败现象的出现,适用于各类型待测snp位点的检测。针对不同的待测snp位点,该第一通用区序列可相同。
另外,对于步骤b中,含待测snp位点的扩增产物与接头的连接,以下将从多个方面进行进一步阐述。
在本发明的一个实施例中,含待测snp位点的扩增产物中仅有一端与接头连接,进而形成待测序文库分子。该连接端可为含待测snp位点的扩增产物的任一端。
在本发明的另一个实施例中,含待测snp位点的扩增产物的两端均与接头连接,进而形成待测序文库分子。
需要说明的是,两端连接的接头的序列可相同或不同。
优选的,两端连接的接头的序列不同,这样可基于这两个不同的接头对待测序文库分子进行扩增验证,验证库分子结构是否正确,也可基于这两个不同的接头对待测序文库分子进行单分子扩增,然后对单分子扩增产物进行第二代高通量基因测序,从而获得待测snp位点的序列信息。
基于上述任一种实施例,本发明提出另一实施例,所述步骤b为:
将含待测snp位点的扩增产物直接与接头连接,形成待测序文库分子,并通过微球可寻址的固定在固相载体上。
本方案中,含待测snp位点的扩增产物直接与接头连接,可有效减少实验步骤,提高实验效率。
需要说明的是所述步骤b可有多种实施方案,以下将通过多个实施例来进行说明。
在本发明的一个实施例中,所述步骤b包括以下步骤:
b1、将含待测snp位点的扩增产物直接与固定在微球上的接头连接,形成固定在微球上的待测序文库分子;
b2、将步骤b1所得微球可寻址的固定在固相载体上。
在本发明的一个实施例中,所述步骤b包括以下步骤:
b1、将将含待测snp位点的扩增产物直接与接头连接,然后将连接产物固定在微球上,得固定在微球上的待测序文库分子;
b2、将步骤b1所得微球可寻址的固定在固相载体上。
需要说明的是,在上述两个实施例中,所述接头上含有修饰标记,用于使其固定在微球上。所述修饰标记可为生物素、亲和素、链霉亲和素、抗原、抗体、受体、配体、多聚组氨酸、纳米金、碘乙酰、巯基、氨基、醛基、羧基、异硫氰基、硅烷基或丙烯酰胺,它们均能特异性的与相对应的基团或分子结合。
所述可寻址的固定,是指能够确定位置信息的固定。即,固定载体上每一具体位置上所固定的待测序文库分子与其它具体位置上所固定的待测序文库分子是能够明确区分的。
上述实施例中,所述接头可采用多种形式,包括但不限于平末端接头、突出末端接头、分叉接头或含茎环结构的接头。
所述平末端接头是指双链之间完全互补配对的双链核酸接头。优选的,所述平末端接头的两条链的5’末端均不含磷酸基团,其可避免连接过程中接头自连现象的出现,减少对后续测序实验的干扰,提高基因检测的准确率。
所述突出末端接头,是指双链核酸分子中的至少一端带有突出核苷酸序列,而其余核苷酸则完全互补的双链核酸接头。突出末端接头可为单突出末端,也可以是含有两个突出末端的双突出末端,这两个突出末端可在一条核苷酸链上或在不同的核苷酸链上。所述突出末端接头能够避免连接过程中接头自连现象的出现,减少对后续测序实验的干扰,提高基因检测的准确率。在本实施例的具体实施方式中,所述接头优选为单突出末端接头,且该突出末端为其所在链的3’末端,碱基为t;该接头能够与通过taq酶扩增的含a尾的pcr扩增产物直接连接,提高连接效率。
所述分叉型接头包括互补区和分叉区,互补区双链的核苷酸互补配对,配对的核苷酸对数不限。互补区末端可为平末端或突出末端。所述分叉型接头能够避免连接过程中接头自连现象的出现,减少对后续测序实验的干扰,提高snp分型检测的准确率。在本实施例的具体实施方式中,所述接头优选为互补区的3’末端为突出末端,且突出末端最后一个碱基为t的t末端分叉接头;该接头能够与通过taq酶扩增的含a尾的pcr扩增产物直接连接,提高连接效率。
所述带茎环结构的接头,有多种实施方案。在一实施例中,该接头为单链核酸分子,该单链核酸分子依次包括第一互补配对区、茎环区和第二互补配对区,第一互补配对区能够与第二互补配对区完全互补配对。在另一实施例中,带茎环结构的接头还可带有突出末端,该突出末端可位于单链核酸分子的3’端。突出末端4的存在能够防止接头自连现象的发生,减少对后续测序实验的干扰,提高snp分型检测的准确率。该突出末端优选为t;该接头能够与通过taq酶扩增的含a尾的pcr扩增产物直接连接,提高连接效率。
上述两个实施例中,接头被预先固定在微球上的实施例减少了连接产物与微球连接的步骤,实验效率更高。
因为固定有接头的微球的密度可能会与步骤b反应体系中的其他成分的密度不同,所以,在连接过程中,使整个反应体系周期性震荡,可有效提高固定在微球上的接头与步骤a所得扩增产物的连接效率,避免反应体系中各成分分层而引起的连接效率低现象的出现。
另外,在本发明中,因为步骤a所得的含待测snp位点的扩增产物直接与步骤b中的接头连接,即不经纯化,直接进行连接反应,这使得反应体系较大,dna分子浓度较低,连接效率不高。在本发明的一个优选实施例中,在所述步骤b的反应体系中加入peg。
因为peg既能够提高反应体系中发生反应的分子的有效密度,提高接头与相应的扩增产物之间接触的概率;又能提高反应体系的密度,防止固定有接头的微球沉降;本方案从两个方面有效提高了固定在微球上的接头与相应的扩增产物的连接效率。本方案中,peg的具体浓度可根据所述步骤b中微球的密度和原反应体系的密度来计算,以使微球的密度与加入peg后的反应体系的密度基本相同为宜。
基于上述实施例,本发明又提出一实施例,所述步骤b1中的连接反应是在切割-连接反应体系中进行的,所述切割-连接反应体系包括:连接酶、断裂剂、固定在微球上的第一接头和连接缓冲液;所述第一通用区中含有可断裂位点;所述可断裂位点能被切割剂特异性切割,使得基于第一引物对获得的扩增产物形成第一粘性末端;所述第一接头为核酸分子,含有与第一粘性末端完全互补配对的第二粘性末端。
本方案中,基于第一通用区中的可断裂位点,扩增产物被断裂剂切出第一粘性末端后,可与含有第二粘性末端的第一接头直接连接,提高连接效率。
优选的,所述可断裂位点为核糖核苷酸、rna序列或限制性内切酶酶切位点。相应的,所述断裂剂为rnaseh、rnaseh或限制性内切酶。
当所述可断裂位点为u时,所述断裂剂优选为user酶。
为了更好的实现本发明的目的,本发明提出第六典型实施例,一种基因测序方法,包括以下步骤:
a、利用上述任一种引物组,对待测样本进行pcr扩增,得含待测snp位点的扩增产物;
b、将含待测snp位点的扩增产物与接头连接,形成待测序文库分子;
c、利用上述任一种锚定引物,对待测序文库分子进行测序,获得待测snp位点的序列信息。
需要说明的是,上述步骤a、b为文库构建步骤,其可采用上述的任一种文库构建方法进行文库构建。以下主要通过对步骤c进行进一步阐述说明。步骤c中的测序方法可为连接测序法也可为合成测序法。
本方案在测序过程中,锚定引物能准确锚定在目标位置上,避免因为待测snp位点附近序列的特殊性而导致的测序结果不准确或测序失败现象的出现,又使得各锚定引物与目标位置结合情况更为一致、归一化,减小因为各锚定引物与目标位置结合情况的差异,从而减小各待测snp位点检测信号的差异,提高测序的准确性。
在本发明的一个实施例中,所述步骤c中的测序方法为连接测序法,所述步骤b、c之间还包括步骤d,采用权利要求7至11中的任一种锚定引物,利用无荧光标记的寡核苷酸探针替换有荧光标记的寡核苷酸探针,对待测序文库分子进行一次连接测序反应。
需要说明的是,所述无荧光标记的寡核苷酸探针与连接测序技术中所使用的有荧光标记的寡核苷酸探针相比,序列完全相同,差别仅在于有无荧光标记;即,所述无荧光标记的寡核苷酸探针序列为(n-n-n……-n)n,所述n为a、g、c或t,n为正整数。
每一次连接测序反应均包括以下步骤:锚定引物锚定,冲洗(除去多余的未锚定引物),探针连接,冲洗(除去多余探针、连接酶等),采图(获得探针上荧光标记所对应位置的序列信息),变性洗脱连接产物(以便下一次连接测序反应中锚定引物的锚定)。步骤c中的连接测序反应,因为使用的是无荧光标记的寡核苷酸探针,因此可不进行采图步骤,以减少实验步骤,提高实验效率。当然,如果进行采图步骤,可验证该次使用的荧光探针是否是无荧光标记的,起到一个再次确认的效果。
虽然,从理论上来说,步骤d中锚定引物与无荧光标记的寡核苷酸案子会被洗脱去除,无法起到封闭的作用,但是,本申请发明人在具体实验中对比发现,在连接测序前,增加一个使用无荧光标记的寡核苷酸探针替换有荧光标记的探针进行一次连接测序反应的步骤,有效的减少在后续的连接测序中出现的错误信号,从而减少由测序初始结果到最终的序列信息的分析过程中的干扰信号,提高snp位点检测的准确性。具体原因可能是非锚定引物的目标结合位点和/或非探针的目标结合位点经过步骤d后被封闭,使得在后续的连接测序实验中,锚定引物和/或探针能够更多更准确的结合在目标结合位点上,从而减少了错误信号的产生,从而提高基因检测过程中正确信号的比例,提高了对snp位点进行检测的准确性。
优选的,所述n在6-10之间;更优选在7-9之间。
更优选的,所述无荧光标记的寡核苷酸探针与步骤d中的有荧光标记的寡核苷酸探针序列完全相同,差别仅在于有无荧光标记。本方案可有效降低无荧光标记的寡核苷酸探针的设计难度。
在本发明的一个实施例中,所述步骤d包括以下步骤:
d1、将所述锚定引物锚定在待测序文库分子上;
d2、加入无荧光标记的寡核苷酸探针、连接酶以及相应的缓冲液,进行连接反应;
d3、变性去除连接产物。
对于步骤d3中变性去除连接产物需要说明的是,步骤d3所述连接产物为锚定引物与无荧光标记的寡核苷酸探针的连接产物,为单链核酸分子,可与待测序文库分子互补配对。所述变性既可以通过物理的方法实现,例如提高双链核酸分子(待测序文库分子与连接产物)所处的温度,也可以通过化学的方法实现,例如改变双链核酸分子(待测序文库分子与连接产物)所处的ph值。其中,采用化学方法,只需加入酸性或碱性的试剂即可,无需额外的加热部件,能够更简单有效的实现自动化。
优选的,所述步骤d3为:用0.05m-0.15mnaoh冲洗。
针对上述各技术方案,为进一步说明本发明所记载技术方案的技术效果及优越性,本发明给出下述具体实施例。
在第一具体实施例中,以10个正常人的血液基因组dna为模板,同时检测cyp2c9基因*2(rs1799853)位点、aldh2基因rs671位点、adrb1基因rs1801253位点。
针对这些位点,分别设计了一对内引物和一对外引物。具体如下所述:
rs1799853内引物对为:seqidno:1和seqidno:2;rs1799853外引物对为:seqidno:3和seqidno:4;rs671内引物对为:seqidno:5和seqidno:6;rs671外引物对为:seqidno:7和seqidno:8;rs1801253内引物对为:seqidno:9和seqidno:10;rs1801253外引物对为:seqidno:11和seqidno:12。
一、含待测snp位点的扩增产物的获得。
1、第一次pcr扩增
以血液基因组dna为模板,利用上述的外引物对,分别对各snp位点所在序列进行扩增,反应体系为:f引物(10μm),0.4μl;r引物(10μm),0.4μl;dntp(各2.5mm),2μl;血液基因组dna,50ng;extaq(5u/μl),0.1μl;10×extaqbuffer,2μl;ddh2o加至20μl。
pcr反应条件如下:94℃5min;94℃20s,57℃20s,72℃25s;重复30个循环;72℃3min。
所得产物即为第一次pcr扩增产物。
2、第二次pcr扩增
以第一次pcr扩增产物为模板,利用上述的内引物对,分别对各snp位点所在序列进行扩增,反应体系为:f引物(10μm),0.4μl;r引物(10μm),0.4μl;dntp(各2.5mm),2μl;第一次pcr扩增产物,0.2μl;extaq(5u/μl),0.1μl;10×extaqbuffer,2μl;ddh2o加至20μl。
pcr反应条件如下:95℃5min;94℃20s,40℃20s,57℃25s;重复5个循环;94℃20s,57℃20s,72℃25s;重复30个循环;72℃3min。
所得产物即为第二次扩增产物。
第二次扩增完成后,经琼脂糖凝胶电泳检测,所有样本的第二次扩增产物中均含有目标分子,且凝胶电泳图显示无杂带,条带单一;说明第二次pcr扩增成功获得了所需的pcr扩增产物。
二、待测序文库分子的构建。
1、将第一接头固定在微球上。
为了在同时检测过程中能够区分不同的样本来源,本具体实施例在第一接头上设计了标签序列,第一接头的基本结构如图4所示(seqidno:13、seqidno:14),该接头中的nnnn为标签序列,标签序列与样本来源和snp位点的对应关系如图5所示。
将上述各第一接头分别与带有链霉亲和素修饰的myone磁珠(invitrogen)结合,使得上述各第一接头被分别固定在磁珠表面,反应体系及反应过程为:将200ng第一接头与4μl(约4×107个磁珠)myone磁珠螺旋振荡混匀,反应30min,以适量te缓冲液(10mmtris-hcl,ph8.0;1mmedta)清洗两次,离心分离,将得到的磁珠以4μl结合缓冲液(10mmtris-hcl,ph7.5;1mmedta;1mnacl;0.01%tritonx-100)重悬保存,得固定有第一接头的磁珠。
2、第二次pcr扩增产物与固定在磁珠上的第一接头的连接。
按按表2中的对应关系分别配置下述反应体系:步骤一所得扩增产物20μl;user酶(1u/μl,neb,cat#m5505s),10μl;缓冲液,8μl;t4dna连接酶,2μl;固定有第一接头的磁珠,0.4μl;加ddh2o至40μl。
其中,所述缓冲液为含400mmtris、100mmmgcl2、100mmdtt、5mmatp、25%peg6000,ph值7.8的溶液。
25℃反应20分钟,得连接产物,即待测序文库分子。
反应结束后,将30管连接产物合并成1管,2500g离心3min,磁铁吸附磁珠,去上清,用50μlte洗涤二次,并最终悬浮于50μlte中,从而将待测序文库分子固定在磁珠上。
3、固定在磁珠上的待测序文库分子的可寻址固定。
将步骤2所得产物点样至异硫氰基修饰的载样片(玻片),于37℃固定1h,即完成固定在磁珠上的待测序文库分子的可寻址固定。
三、测序。
对步骤二所得的固定在固相载体上的待测序文库分子进行测序。
在本具体实施例中采用深圳华因康基因科技有限公司的高通量基因测序仪pstariia测序仪,并采用连接测序法进行测序。测序过程中,
针对rs1799853位点,所使用的锚定引物为seqidno:15;针对rs671位点,所使用的锚定引物为seqidno:16;针对rs1801253位点,所使用的锚定引物为seqidno:17。
另外,为了区分不同的样本来源,还需对第一接头上的标签序列进行检测,用于检测标签序列的锚定引物为seqidno:18。
需要说明的是,seqidno:15-18的的5’端均被磷酸化修饰。
整体的测序顺序如图6所示。其中,待测snp位点anchor混合物为seqidno:15-17这3种锚定引物按相同摩尔数混合所得的混合物。其中,base1用于封闭锚定引物或探针与待测序文库分子上的非特异性结合位点,base2用于检测待测snp位点,base3-6用于检测标签序列。
另外,本实施例做了第一对比实验,同样步骤二所得产物,以图7中的测序顺序进行测序。即,未进行图6中的base1。
图8示出了上述具体实施例中样本2的待测snp位点——rs1799853、rs671、rs1801253在实验组和第一对比组中的初步实验结果。其中,r=a/g,y=c/t,m=a/c,k=g/t,s=c/g,w=a/t,h=a/c/t,b=c/g/t,v=a/c/g,d=a/g/t,n=a/c/g/t;n表示信号太弱,无法判断是a、c、g和t中的哪一个。
由实验结果可知,相同样本在实验组和第一对比组中占比最高的snp位点序列类型均相同,但是它们的占比有明显区别;实验组中占比最高的snp位点序列类型检出数均在95%以上,而第一对比组中占比最高的snp位点序列类型检出数基本在85%左右。
另外,本发明人还将第二次扩增产物通过sanger测序进行验证,作为第二对比实验,结果显示,上述实验组和第一对比组中占比最高的snp位点序列类型均与sanger测序结果完全一致。
为了证明本发明的引物组、锚定引物的技术效果,本实验发明人还做了第三对比实验,同样以上述具体实施例中的正常人的血液基因组dna为模板,同时检测cyp2c9基因*2(rs1799853)位点、aldh2基因rs671位点、adrb1基因rs1801253位点。
针对这些位点,分别设计了一对内引物和一对外引物。具体如下所述:
rs1799853内引物对为:seqidno:19和seqidno:20;rs1799853外引物对为:seqidno:3和seqidno:4;rs671内引物对为:seqidno:21和seqidno:22;rs671外引物对为:seqidno:7和seqidno:8;rs1801253内引物对为:seqidno:23和seqidno:24;rs1801253外引物对为:seqidno:11和seqidno:12。
一、含待测snp位点的扩增产物的获得。
1、第一次pcr扩增。
以血液基因组dna为模板,利用上述的外引物对,分别对各snp位点所在序列进行扩增,反应体系为:f引物(10μm),0.4μl;r引物(10μm),0.4μl;dntp(各2.5mm),2μl;血液基因组dna,50ng;extaq(5u/μl),0.1μl;10×extaqbuffer,2μl;ddh2o加至20μl。
pcr反应条件如下:94℃5min;94℃20s,57℃20s,72℃25s;重复30个循环;72℃3min。
所得产物即为第一次pcr扩增产物。
2、第二次pcr扩增。
以第一次pcr扩增产物为模板,利用上述的内引物对,分别对各snp位点所在序列进行扩增,反应体系为:f引物(10μm),0.4μl;r引物(10μm),0.4μl;dntp(各2.5mm),2μl;第一次pcr扩增产物,0.2μl;extaq(5u/μl),0.1μl;10×extaqbuffer,2μl;ddh2o加至20μl。
pcr反应条件如下:95℃3min;94℃20s,57℃20s,72℃30s;重复30个循环;72℃3min。
所得产物即为第二次扩增产物。
第二次扩增完成后,经琼脂糖凝胶电泳检测,所有样本的第二次扩增产物中均含有目标分子,且凝胶电泳图显示无杂带,条带单一;说明第二次pcr扩增成功获得了所需的pcr扩增产物。
二、待测序文库分子的构建。
采用上一具体实施例相同的方法构建测序文库并将待测序文库分子可寻址固定。
三、测序。
对步骤二所得的固定在固相载体上的待测序文库分子进行测序。
在第三对比实验中同样采用深圳华因康基因科技有限公司的高通量基因测序仪pstariia测序仪,并采用连接测序法进行测序。测序过程中,针对rs1799853位点,所使用的锚定引物为seqidno:25;针对rs671位点,所使用的锚定引物为seqidno:26;针对rs1801253位点,所使用的锚定引物为seqidno:27。
另外,为了区分不同的样本来源,还需对第一接头上的标签序列进行检测,用于检测标签序列的锚定引物为seqidno:18。
需要说明的是,seqidno:18、25-27的5’端均被磷酸化修饰。
然后参考上一具体实施例中的方法,更换锚定引物后,按照图6的顺序进行测序;另外,同样以本具体实施例步骤二所得产物,更换锚定引物后,按照图7的顺序进行测序,作为对照。
其中,rs671和rs1801253位点的测序采图结果如图9(实验组和对照组)所示,由图9可知,这两个位点在两次测序过程中均无荧光发生,测序失败;这与上一具体实施例中rs671和rs1801253的测序采图结果(图10实验组和对照组)存在明显区别。而rs1799853位点的实验组测序采图结果如图11所示,与上一具体实施例中的实验组rs1799853位点的测序采图结果(图12)相比,同样存在明显区别,荧光信号明显较弱。rs1799853位点的对照组测序采图结果与上一具体实施例中对照组的测序采图结果,与实验组类似,荧光信号明显较弱。
经本发明人分析,rs671和rs1801253位点的在本具体实施例中,实验组和对照组均测序失败的原因可能是:rs671和rs1801253这两个位点附近的序列中存在复杂结构,导致锚定引物无法锚定,进而无法实现锚定引物与探针的连接,所以采图时无信号。
而rs1799853位点在本具体实施例中实验组和对照组的测序采图结果分别与上一具体实施例中的实验组和对照组的测序采图结果相比,荧光信号都明显较弱。原因可能是本具体实施例中,锚定引物与待测序文库分子的结合效果,较上一具体实施例差。
在本发明的第二具体实施例中,直接用上述实施例中的rs1799853内引物对为:seqidno:1和seqidno:2;rs671内引物对为:seqidno:5和seqidno:6;rs1801253内引物对为:seqidno:9和seqidno:10。对上述样本1-10进行pcr扩增,然后进行后续的步骤二、三,所得实验结果和第一具体实施例结果基本一致,实验组中占比最高的snp位点序列类型检出数均在93%以上,而对照组中占比最高的snp位点序列类型检出数基本在80%左右。整体来说,实验组和对照组的检出数和检出总数均较上一实施例低,且无论是在实验组还是在对照组中,占比最高的snp位点序列类型检出数所占比例有所降低。
需要说明的是,上述具体实施例中,所述使用的引物中,可断裂位点均为u,且均设计在上游引物上,断裂序列(与第一粘性末端对应)为cggu。当然,也可将可断裂位点设计在下游引物上。所述断裂序列可根据需要进行设计,只要能满足后续的连接实验的要求即可,例如:gccu、aaau、cgcu、gcccu、cggcu、ttttu等。随着所述断裂序列的改变,所述第一接头的相关序列需进行相关调整。
另外,上述具体实施例中,测序文库分子构建完成后,均未采用单分子扩增技术进行扩增,而是直接进入到后续的测序步骤,简化了实验步骤。当然,本申请发明人经过实验发现,通过单分子扩增技术对待测序文库分子进行扩增后再进行测序,所得实验结果与上述实验结果基本相同。相对来说,测序采图所得信号会更高一点。
以上,本发明的方法能够高效的同时对多个样品的多个snp位点同时进行检测,并得到准确可信的结果,并且能够有效的降低连接测序所得结果中的错误信号,提高snp位点检测的准确性。
应当说明的是,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
sequencelisting
<110>盛司潼
<120>引物组、锚定引物、试剂盒、文库构建及基因测序方法
<130>
<160>27
<170>patentinversion3.3
<210>1
<211>26
<212>dna
<213>人工序列
<400>1
cgguctcagcctcccaagttgaggac26
<210>2
<211>21
<212>dna
<213>人工序列
<400>2
gtgagaaacagcacacaagtt21
<210>3
<211>20
<212>dna
<213>人工序列
<400>3
agcttcctctttcttgcctg20
<210>4
<211>21
<212>dna
<213>人工序列
<400>4
tgagctaacaaccaggactca21
<210>5
<211>26
<212>dna
<213>人工序列
<400>5
cgguctcagcctcccaagcatacact26
<210>6
<211>21
<212>dna
<213>人工序列
<400>6
gtgagaaacagcattttcact21
<210>7
<211>20
<212>dna
<213>人工序列
<400>7
aacccataacccccaagagt20
<210>8
<211>21
<212>dna
<213>人工序列
<400>8
cagcaggtcctgaacttccag21
<210>9
<211>26
<212>dna
<213>人工序列
<400>9
cgguctcagcctcccaagccttccag26
<210>10
<211>21
<212>dna
<213>人工序列
<400>10
gtgagaaacagcaagagcagt21
<210>11
<211>22
<212>dna
<213>人工序列
<400>11
tctgctggctgcccttcttcct22
<210>12
<211>22
<212>dna
<213>人工序列
<400>12
acgacatcgtcgtcgtcgtcgt22
<210>13
<211>50
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(23)..(26)
<223>nisa,c,g,ort
<400>13
cctccctgcagtctctatgggcnnnnctgctagtcgctgaagtagtcggt50
<210>14
<211>42
<212>dna
<213>人工序列
<400>14
ctacttcagcgactagcagacgtgcccatagagactgcaggg42
<210>15
<211>17
<212>dna
<213>人工序列
<400>15
gtcctcaacttgggagg17
<210>16
<211>17
<212>dna
<213>人工序列
<400>16
agtgtatgcttgggagg17
<210>17
<211>17
<212>dna
<213>人工序列
<400>17
ctggaaggcttgggagg17
<210>18
<211>17
<212>dna
<213>人工序列
<400>18
gcccatagagactgcag17
<210>19
<211>22
<212>dna
<213>人工序列
<400>19
cgguctcatgacgctgcggaat22
<210>20
<211>19
<212>dna
<213>人工序列
<400>20
gatatggagtagggtcacc19
<210>21
<211>26
<212>dna
<213>人工序列
<400>21
cgguactgctatgatgtgtttggagc26
<210>22
<211>20
<212>dna
<213>人工序列
<400>22
agcaggtcccacactcacag20
<210>23
<211>22
<212>dna
<213>人工序列
<400>23
cggugtgcccgaccgcctcttc22
<210>24
<211>18
<212>dna
<213>人工序列
<400>24
gtctccgtgggtcgcgtg18
<210>25
<211>17
<212>dna
<213>人工序列
<400>25
gtcctcaatgctcctct17
<210>26
<211>17
<212>dna
<213>人工序列
<400>26
agtgtatgcctgcagcc17
<210>27
<211>17
<212>dna
<213>人工序列
<400>27
ctggaaggccttgcgga17
- 还没有人留言评论。精彩留言会获得点赞!