双顺反子细菌表达系统的制作方法
本发明涉及用于产生目的蛋白质的单一载体中的双独立顺反子表达系统,所述目的蛋白质包含表达为在细菌大肠杆菌(E.coli.)中形成的不溶性包涵体的抗体的重组Fab片段或其它抗体片段、肽和蛋白质。
背景技术:
重组DNA技术(rDNA)已经彻底改变了治疗剂的制备方式。所需蛋白质现在在外源细胞内制备并纯化。
具有翻译后修饰(PTM)的蛋白质通常在哺乳动物或酵母系统中表达为重组分子。酵母表达系统如毕赤酵母属(Pichia)和酵母属(Saccharomyces)在PTM方面更接近哺乳动物系统,但是在糖基化类型中依然不同,如在毕赤酵母属情况下的高甘露糖聚糖,使得其不适合用于人使用的重组蛋白质的表达。
单克隆抗体(mAb)、抗体、融合蛋白、mAb的Fab片段被用作治疗剂。rDNA技术使用专门的载体和表达系统来产生治疗性蛋白质。表达系统主要由细菌、酵母、昆虫或哺乳动物表达系统组成。最初,大多数重组蛋白质在使用大肠杆菌作为宿主的细菌表达系统中表达。使用大肠杆菌作为表达宿主具有多个优点,例如容易克隆、容易表达、更短的时间表、更短的孵育期和非常高的产量。因此,可以在大肠杆菌中安全地表达不需要任何PTM的蛋白质。
作为mAb的抗原结合片段部分的Fab不需要在哺乳动物系统中表达,因为其不包含存在于抗体Fc部分中的糖基化位点。因此,Fab通常在大肠杆菌系统中表达。在1980-1990期间,数位研究人员尝试了在大肠杆菌中表达Fab。PlückthunA等人,1990Behring Inst.Mitt.(87):48-55是报道了从大肠杆菌中分泌Fab抗体的一些早期工作人员。Williamson R.A.等人,1991Biochem J.277(Pt 2):561-3报道了使用噬菌体λ载体在大肠杆菌中表达Fab分子。用于在大肠杆菌中生产Fab、二价抗体或嵌合抗体片段的噬菌体展示系统。此外,Fab也在大肠杆菌中作为错误折叠的包涵体产生,然后将其重折叠以获得功能分子,从而获得抗体产率40%的提高。
上述大多数研究使用单启动子(即phoA)来驱动重链和轻链两者的表达。存在于重链和轻链之间的核糖体结合位点(rbs)驱动第二基因的转录和翻译。
US5648237也采用类似的单启动子(phoA)策略来在大肠杆菌中表达Fab基因,以获得分泌产物。上述策略的主要缺点是第二基因的表达水平通常低于第一基因,因此限制功能性Fab的产量。
专利No.WO03018771公开了通过两个分开的翻译单元产生抗体的方法,所述翻译单元分别编码所述抗体或片段的轻链和重链,其中两条链以顺序方式表达,从而特异性地分离轻链和重链的产生,并允许轻链和重链组装。
专利No.EP1356052B1公开了在原核细胞中产生全抗体的方法。存在第一启动子和第一顺反子以产生免疫球蛋白轻链,以及第二启动子和第二顺反子以产生免疫球蛋白重链,其中两条链折叠并组装以形成生物活性免疫球蛋白。
技术实现要素:
在一个实施方案中,本发明涉及单一载体中的双独立顺反子表达系统,其用于在细菌细胞中产生表达为不溶性包涵体的重组蛋白质和肽。
在另一个实施方案中,本发明涉及制备具有两个不同启动子的单一载体中的双独立顺反子表达系统的方法,其用于在细菌细胞中产生表达为不溶性包涵体的重组蛋白质和肽。
在另一个实施方案中,本发明涉及具有两个不同启动子的单一载体中的双独立顺反子表达系统,其用于在细菌细胞中产生表达为不溶性包涵体的抗体片段。
在另一个实施方案中,本发明涉及具有两个不同启动子的单一载体中的双独立顺反子表达系统,其用于在细菌细胞中产生表达为不溶性包涵体的抗体的重组Fab片段。
在另一个实施方案中,本发明涉及具有两个不同启动子的单一载体中的双独立顺反子表达系统,其用于在细菌细胞中产生表达为不溶性包涵体的重组肽。
在另一个实施方案中,该双顺反子表达系统包含:
a)第一顺反子,其包含与编码目的蛋白质的多核苷酸序列可操作地连接的启动子;
b)第二顺反子,其包含与编码目的蛋白质的多核苷酸序列可操作地连接的启动子;
其中所述第一顺反子和所述第二顺反子位于单一载体中,并在细菌细胞中表达编码作为包涵体的目的蛋白质的多核苷酸序列。
在另一个实施方案中,本发明涉及双顺反子载体,其包含可操作地连接到含有目的基因的多克隆位点的启动子、核糖体结合位点和终止子。
在另一个实施方案中,本发明涉及使用双顺反子表达系统产生目的蛋白质的方法。
下面阐述的本发明的一个或多个实施方案的细节在本质上仅仅是说明性的,并且不旨在限制本发明的范围。通过该描述,本发明的其它特征、目的和优点将变得明显。
附图简述
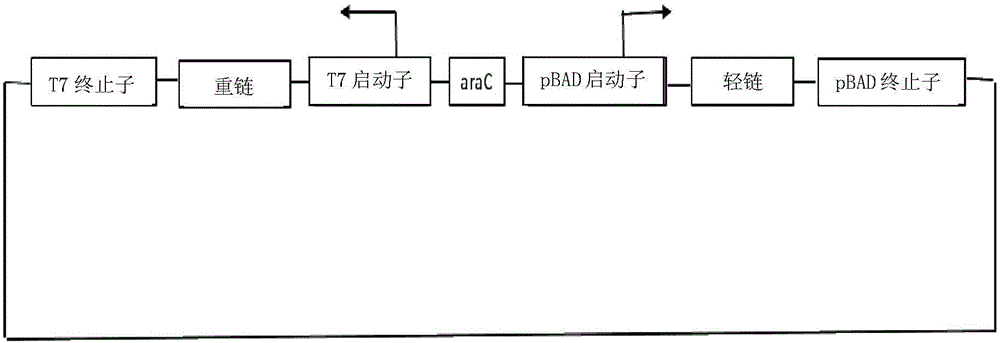
图1;示出了双顺反子载体的公式。
图2;示出了克隆pET21a-HC-LC的载体图谱。
图3;示出了大肠杆菌BL21A1克隆的不溶性沉淀部分以及对照和参考产品的SDS PAGE分析。
图4;示出了溶解的IB样品的RP-HPLC分析,与还原的Fab分子相比,在克隆中看到了LC和HC峰。
图5;示出了将重链峰与其它蛋白质分离而运行的HPLC。
图6;示出了与在大肠杆菌BL21A1细胞系中的单顺反子克隆相比,双顺反子构建体中SAK-Lira克隆的表达显著增加。
图7;示出了载体图谱pBAD24M-LC。
发明详述
定义:
如本文所用,术语“目的蛋白质”在本文中是指用于生物治疗行业或用于诊断或研究目的的任何多肽,包括蛋白质和肽。
如本文所用,本文所用的术语“编码目的蛋白质的多核苷酸序列”包括编码表达多肽的基因(优选异源基因)的DNA。
如本文所用,术语“重组蛋白质和肽”是指在活细胞内通过重组DNA的表达产生的蛋白或肽。
如本文所用,术语“Fab”和“抗体”可互换使用,因为抗体包含两个部分,即Fab和Fc区。
如本文所用,术语“载体(vector)”是指用作人工携带外源遗传物质进入细菌细胞的媒介物(vehicle)的DNA分子,其可以在细菌细胞中复制和表达。
如本文所用,术语“顺反子”是指含有单个多肽的遗传密码并用作遗传单位的DNA区段。
如本文所用,术语“双独立顺反子表达”是指两个分开的顺反子,其用于独立地表达两种相同或不同的蛋白质。
如本文所用,术语“相同(same)”与同一(identical)或相似(similar)可互换。
如本文所用,术语“双顺反子表达系统”包含编码待表达的多肽的多核苷酸序列和控制其表达的序列,例如启动子和任选的增强子序列。本发明的启动子可操作地连接到待表达的基因(即转录单位),或通过插入DNA(例如通过异源基因的5'非翻译区)与其分开。优选地,表达系统的侧翼是一个或多个合适的限制性位点,以便能够将表达盒插入载体和/或将其从载体中删除。因此,根据本发明的表达系统可用于构建表达载体,特别是细菌表达载体。
如本文所用,术语“启动子”是指通常位于基因上游的DNA调节区,提供用于调节基因转录的控制点。
如本文所用,术语“可操作地连接”是指两个或更多个DNA区段,特别是待表达的基因序列和控制其表达的那些序列之间的功能关系。
如本文所用,术语“小肽”或“肽”是指用于生物治疗行业以及诊断和研究目的的2至10kDa的肽,如利拉鲁肽、exanetide、PTH等。
本发明提供了用于产生多种目的重组蛋白质的双顺反子表达系统。在某些实施方案中,双顺反子表达系统包含两个顺反子,其具有与编码目的蛋白质的多核苷酸序列可操作地连接的启动子和终止子。
在某些实施方案中,双顺反子表达系统包含位于单一载体中的两个顺反子,其表达编码目的蛋白质的多核苷酸序列。
在一个实施方案中,双顺反子表达系统包含:
a)第一顺反子,其包含与编码目的蛋白质的多核苷酸序列可操作地连接的启动子;
b)第二顺反子,其包含与编码目的蛋白质的多核苷酸序列可操作地连接的启动子;
其中所述第一顺反子和所述第二顺反子位于单一载体中,并表达编码作为宿主细胞中形成的包涵体的目的蛋白质的多核苷酸序列。
在一个实施方案中,启动子可以选自T7启动子、阿拉伯糖启动子phoA、tac、lpp、lac-lpp、lac、trp、trc,优选T7启动子和阿拉伯糖启动子。在某些实施方案中,双顺反子表达系统包含两个顺反子,其在两个启动子的控制下表达编码目的蛋白质的多核苷酸序列。在一个实施方案中,两个启动子控制编码相同目的蛋白质的多核苷酸序列的表达。在另一个实施方案中,两个启动子控制编码氨基酸长度或物理化学性质不同的目的蛋白质的多核苷酸序列的表达。
在某些实施方案中,目的蛋白质可以选自肽和蛋白质。
在一些实施方案中,蛋白质可以在双顺反子载体中表达。所述蛋白质包括抗体或其片段。抗体片段可以在双顺反子表达系统中表达。抗体片段可以选自抗体的Fab重链和轻链或其它抗体片段,例如scFv、双抗体、三抗体、四抗体、双-scFv、微抗体Fab2(双特异性)、Fab3(三特异性)。在优选的实施方案中,双顺反子表达系统表达编码形成Fab抗体的抗体重链和轻链的多核苷酸序列。在这样的实施方案中,Fab抗体显示对VEGF受体的亲和力,并且所述Fab抗体是雷珠单抗(Ranibizumab)。
在另一个实施方案中,蛋白质可以选自但不限于G-CSF、IFN、促红细胞生成素、胰岛素及其变体、PTH(1-84aa)、FSH、LH、GH和蛋白质二硫化物异构酶(PDI)。
在一些实施方案中,所述肽可以在双顺反子载体中表达。所述肽包含选自至少小于40个氨基酸或优选小于31个氨基酸或更优选小于10个氨基酸的氨基酸序列。在某些实施方案中,肽分子量选自约2至约10kDa。所述肽可以选自但不限于GLP-1肽类似物如利拉鲁肽或Exendinor GLP-2肽如替度鲁肽和PTH(1-34aa)和胰岛素。在另一个优选的实施方案中,双顺反子表达系统表达编码GLP-1激动剂肽的多核苷酸序列。在这样的实施方案中,GLP-1肽是利拉鲁肽。
在另一个实施方案中,两个启动子独立地控制氨基酸长度和物理化学性质不同的不同目的蛋白质(例如抗体的重链或轻链)的表达。
在一个实施方案中,双顺反子表达系统包含:
a)第一顺反子,其包含与编码抗体重链的多核苷酸序列可操作地连接的T7启动子;
b)第二顺反子,其包含与编码抗体轻链的多核苷酸序列可操作地连接的阿拉伯糖启动子;
其中所述第一顺反子和所述第二顺反子位于单一载体中,并表达作为宿主细胞中形成的包涵体的抗体的重链和轻链。
在这样的实施方案中,抗体的抗体重链和轻链包含核苷酸序列序列ID no.1和序列ID no.2或氨基酸序列序列ID no.3和序列ID no.4。在一些实施方案中,第一顺反子和第二顺反子的位置可互换,其中第二顺反子可以克隆在载体中第一顺反子的位置,第一顺反子可以位于第二顺反子处。抗体的重链和轻链独立地表达为包涵体,并且可以进一步处理以获得对VEGF受体显示亲和力的Fab抗体,并且所述Fab抗体是雷珠单抗。
在某些实施方案中,抗体的重链和轻链任选地与信号肽(优选pelB)组合表达。信号肽指导蛋白在宿主细胞的周质空间中的表达。
在实施方案中,在具有分别调节重组Fab片段的重链和轻链的产生的阿拉伯糖启动子和T7启动子的两个不同启动子且均具有pelB标签的单一载体中的双顺反子表达系统在大肠杆菌的周质空间中产生为不溶性包涵体。
在某些实施方案中,抗体的重链或轻链任选地与调节子(优选AraC基因)组合表达,以进一步增加蛋白质的表达。
双顺反子表达系统提供目的蛋白质的等摩尔表达。为了以合适的质量和数量获得目的蛋白质,非常需要等摩尔表达。其取决于重链与轻链的比例或克隆到载体中的多肽亚基的比例。在某些实施方案中,重链和轻链以合适的比率克隆,包括重链至少等于或高于轻链,以获得重链和轻链的等摩尔表达。以选自1:5:0.7至1:1,包括1:3:0.8、1:2:0.9、1:2:1 1:1的比例克隆重链和轻链。
在实施方案中,双顺反子表达系统包含序列ID no 19所示的核苷酸序列。
在另一个实施方案中,双顺反子表达系统包含:
a)第一顺反子,其包含与编码肽的多核苷酸序列可操作地连接的T7启动子;
b)第二顺反子,其包含与编码肽的多核苷酸序列可操作地连接的阿拉伯糖启动子;
其中所述第一顺反子和所述第二顺反子位于单一载体中,并表达作为宿主细胞中形成的包涵体的所述肽。
在这样的实施方案中,所述肽是GLP-1类似物,包含序列ID no 6所示的核苷酸序列,其编码为具有序列ID No 7的氨基酸序列的利拉鲁肽的GLP-1激动剂肽。
在某些实施方案中,肽可以任选地与本领域技术人员已知的信号肽或调节子/增强子一起表达。
在某些实施方案中,所述肽可任选地与融合伴侣或融合标签一起表达,以防止肽的降解。融合伴侣包含30个氨基酸至300个氨基酸的氨基酸序列。融合伴侣包含选自约50个氨基酸、100个氨基酸、约136个氨基酸、约175个氨基酸、约250个氨基酸、300个氨基酸,优选约136个氨基酸的氨基酸序列。融合标签可以选自但不限于组氨酸标签、谷胱甘肽-s-转移酶(GST)、麦芽糖结合蛋白、NusA、硫氧还蛋白(TRX)、多组氨酸(HIS)、小泛素样修饰剂(SUMO)和泛素(Ub)和葡萄球菌激酶(SAK)基因。在优选的实施方案中,融合标签是SAK基因。SAK基因作为目的蛋白质的融合标签的详细用途公开于US8853380中,其通过引用并入本文。
在一些实施方案中,双顺反子表达系统还包含选择标记,其选自氨苄青霉素、卡那霉素,优选氨苄青霉素。
在另一个实施方案中,本发明提供了产生目的蛋白质的方法,其包括以下步骤:
(i)用基本上由双顺反子表达系统组成的单一载体转化宿主细胞;
(ii)在合适的培养基中培养转化的细胞以表达目的蛋白质,其中第一顺反子和第二顺反子表达在包涵体中的目的蛋白质;
(iii)进行包涵体的溶解;
(iv)进行目的蛋白质的重折叠。
在实施方案中,将双顺反子表达系统转染到合适的细菌宿主细胞中以表达目的蛋白质。合适的细菌宿主细胞是大肠杆菌,其中目的蛋白质以包涵体的形式表达。包涵体是在大肠杆菌的周质或细胞质中形成的不溶性物质。可以通过本领域熟知的技术分离、溶解包涵体,并且以活性形式回收目的蛋白质。
在一个实施方案中,通过在具有两个不同启动子(阿拉伯糖启动子和T7启动子)的单一载体中构建两个独立的顺反子,将抗体的Fab重链和轻链或其它抗体片段,例如scFv、双抗体、三抗体、四抗体、双-scFv、微抗体Fab2(双特异性)、Fab3(三特异性)表达为大肠杆菌的周质空间中的不溶性包涵体。两个不同的启动子,即T7启动子和阿拉伯糖启动子分别有助于Fab分子的重链和轻链的表达。抗体重链和轻链在细菌细胞即大肠杆菌中作为非功能性包涵体产生,随后对其进行提取、重折叠和纯化。
在本发明的一个实施方案中,顺反子包含这样的结构,即,每个基因(重链和轻链)在单个载体中具有其自身的启动子和终止子。在T7启动子的控制下克隆重链,而在阿拉伯糖启动子的控制下克隆轻链。两条链之前是信号序列pelB标签,用于在细菌膜的周质空间中获得的产物。
双顺反子表达系统的优点是,阿拉伯糖和T7两者都是强启动子,从单次发酵运行获得轻链和重链两者的高表达,而不是用轻链和重链克隆分开发酵。双顺反子表达系统使得更容易表征和维持单细胞库,而不是用于轻链和重链克隆的分离的细胞库。此外,当从细菌细胞的周质空间提取时,由此获得的包涵体是相对纯的。作为包涵体获得的高水平的表达和更纯形式的轻链和重链相对更容易在离体折叠成功能性Fab,从而显著地增加产物的产量。
该系统的另一个优点是可以在阿拉伯糖启动子和T7启动子下克隆和表达目的蛋白质,并且可以显著提高蛋白质的表达水平。
下面公开的实施例仅用于说明本发明的目的,而不是限制性的。
实施例1:在pET21a载体中克隆重链
用于克隆Fab片段的重链和轻链的DNA序列分别在序列ID no 1和2中给出。使用基因特异性引物从合成DNA来扩增重链插入物。根据本领域熟知的方法设计引物。然后将重链PCR产物用NdeI-HindIII酶消化,并连接到用相同酶消化的pET21a载体。通过菌落PCR筛选克隆体并通过限制性分析证实。将所得克隆体命名为pET21a-HC。将重组载体导入BL21A1细胞系并检查重链的表达。
实施例2:在pBAD24M载体中克隆轻链
使用基因特异性引物从合成DNA来扩增轻链插入物。根据本领域熟知的方法设计引物。将扩增的轻链用NdeI-HindIII酶消化,并在相同位点连接到消化的pBAD24M载体(实验室中可获得)。通过菌落PCR筛选克隆体并通过限制性分析证实。将所得克隆体命名为pBAD24M-LC。将重组载体导入BL21A1细胞系并检查轻链的表达。
实施例3:在同一载体中构建两个独立的顺反子
设计引物以与阿拉伯糖启动子、终止子和araC基因一起扩增轻链。根据本领域熟知的方法设计引物。引物将BglII接头加入扩增产物。pET21a载体具有在T7启动子上游的单个BglII位点。使用载体特异性引物从模板pBAD24M来扩增轻链表达盒,并在BglII位点克隆到pET21a-HC克隆体中。通过限制性消化和测序确认所述克隆体。最终的克隆体命名为pET21a-HC-LC,并基于表达列出合适的克隆体。pET21a-HC-LC的克隆图谱示于图2中。
这样产生的克隆体含有重链和轻链两者的独立调节和表达所需的所有区段。
实施例4:表达分析
使用大肠杆菌BL21A1细胞系作为表达宿主。除了BL21A1之外,使用BL21DE3或基因组中含有T7启动子的任何其它细胞系。使用上述选择的克隆体连同作为对照的pET21a-HC和pBAD24M-LC转化BL21A1细胞。通过IPTG诱导重链,通过阿拉伯糖诱导轻链。诱导物浓度为13mM阿拉伯糖和1mM IPTG,当培养物OD 600为~1时完成诱导。诱导后4小时收获细胞。在摇瓶中进行研究。将获得的收获物用珠裂解并离心以分离可溶性级分和不溶性级分。将样品装载在12%SDS PAGE凝胶上以检查表达。SDS PAGE凝胶分析示于图3中。将还原的雷珠单抗加载到图3的泳道5中以证实还原的轻链和重链的表达。
SDS PAGE分析显示不溶性沉淀级分中两条链的表达,并且也通过RP-HPLC分析得以证实,其中参考产品的轻链和重链的保留时间对应于内部产物的保留时间。使用的对照是还原的Fab分子(参考产品),以及pET21a-HC克隆体和pBAD24M克隆体的产物。因此,证实了来自单个克隆体的重链和轻链的表达。RP-HPLC分析示于图4和5中。在RP-HPLC中,将双顺反子克隆体的溶解和还原的IB与还原的雷珠单抗(RMP)以及分别表达重链和轻链的克隆体(即,pET21-HC和pBAD24MLC)相比较。
仅表达轻链的pBAD24MLC的溶解的IB的主峰保留时间(RT)与还原的RMP的轻链的RT匹配。在RT 13分钟的杂质峰与重链的保留时间匹配,这表明相似的疏水性。因此,通过LC-MS/MS表征杂质,并且最终注释为在RT 17分钟的宿主细胞蛋白质OMP C和具有未裂解的前导序列的轻链。由pET21a-HC表达的重链与参考标准重链匹配。该图谱还表明在RT 19分钟的后峰,其表征为具有未裂解的前导序列的重链。表达LC和HC两者的双顺反子克隆体pET21a_HC_LC具有2个主峰,其具有与参考标准品的LC和HC相当的保留时间。但是,由于OMP C与重链共洗脱,反相测试方法需要更好地解析,这在图4中给出。
将Zorbax C8RP柱上的现有方法修改为Aeriswidepore C8,并解析共洗脱物质。在Aeriswidepore C8上,pET21a_HC_LC的溶解的IB显示出不同的LC、HC和OMP C峰,使得能够鉴定和精确定量IB中的各个亚基,如图5所示。
虽然上面已经详细描述了某些实施方案和实施例,但是本领域普通技术人员将清楚地理解,在不脱离其教导的情况下,可以对实施方案和实施进行许多修改。
实施例no.5:在pET24a载体中克隆具有葡萄球菌激酶(SAK)融合标签的小肽(利拉鲁肽)
使用基因特异性引物由合成DNA扩增SAK和利拉鲁肽基因。根据本领域中熟知的方法设计引物,将PCR产物用NdeI-BamHI和BamHI-HindIII酶消化并且在NdeI-HindIII位点连接到消化的pET24a载体。通过菌落PCR筛选克隆体并通过限制性分析证实。将所得的克隆体命名为pET24a-SAK-Lira。
实施例6:在pBAD24M载体中克隆具有葡萄球菌激酶融合标签的小肽(利拉鲁肽)
如实施例no 6中所给出的,将具有SAK标签的利拉鲁肽克隆到pBAD24M载体中。将该克隆体命名为pBAD24M-SAK-Lira。
实施例no.7:在同一载体中构建两个独立的顺反子,两个顺反子均表达SAK-Lira融合肽。
使用实施例no.3中使用的克隆设计策略构建利拉鲁肽的双顺反子克隆体,其中从pBAD24M-SAK-Lira克隆体扩增SAK-Lira融合基因以及阿拉伯糖表达盒,并克隆到pET24a-SAK-Lira克隆体中以构建双顺反子构建体。将该克隆体标记为pET-ara-SAK-Lira。
实施例no.8:具有SAK-Lira融合蛋白的双顺反子克隆体的表达分析。
使用大肠杆菌BL21A1细胞系作为表达宿主。除了BL21A1之外,使用BL21DE3或基因组中含有T7启动子的任何其它细胞系。使用上述单和双顺反子构建体转化BL21A1细胞。通过IPTG和阿拉伯糖诱导克隆体。诱导物浓度为13mM阿拉伯糖和1mM的IPTG,当培养物OD 600为~1时完成诱导。诱导后4小时收获细胞。在摇瓶中进行研究。将获得的收获物用珠裂解并离心以分离可溶性级分和不溶性级分。将样品装载在12%SDS PAGE凝胶上以检查表达。
SDS PAGE凝胶分析清楚地显示,与单顺反子pET24a-SAK-Lira克隆体(图6泳道3)相比,在双顺反子克隆体(图6泳道2)中SAK-Lira融合蛋白的表达增加。
- 还没有人留言评论。精彩留言会获得点赞!