环氧烷聚合催化剂和使用其制造聚环氧烷的方法与流程
本发明涉及环氧烷聚合催化剂和使用其制造聚环氧烷的方法,尤其是涉及即使为低温的制造条件也能够高效地制造高分子量、低不饱和度且显示出狭窄的分子量分布的聚环氧烷的新型环氧烷聚合催化剂、以及使用其制造聚环氧烷的方法。
背景技术:
聚环氧烷通过以氢氧化钾作为催化剂并进行环氧烷的加聚来制造在工业上是已知的。然而,通过该方法制造高分子量的聚环氧烷时,所得聚环氧烷包含大量一元醇,因此存在不饱和度变高的课题。
作为聚环氧烷的制造方法,提出了使用复金属氰化物络合物作为催化剂来制造聚环氧烷的方法(例如参照专利文献1、2)。
此外,作为不饱和度低的聚环氧烷的制造方法,提出了将特定的膦腈盐用作催化剂来制造聚环氧烷的方法(例如参照专利文献3~5)。
另外提出了下述方法:使用将含活性氢的化合物与铝氧烷以含活性氢的化合物中的活性氢与铝氧烷中的铝原子的比率达到0.2~1:1(摩尔比)的方式混合而得到的环氧烷聚合催化剂,进行环氧烷的开环聚合,从而制造聚环氧烷(例如参照专利文献6)。
进而,作为聚环氧烷的制造方法,提出了下述方法:使用将含活性氢的化合物、膦腈化合物、三异丁基铝以1:1:2的摩尔比混合得到的环氧烷聚合催化剂,在甲苯溶剂中以20℃进行环氧烷的开环聚合,从而制造聚环氧烷(例如参照非专利文献1)。关于此时的催化活性种,提出了下述机理:首先,通过含活性氢的化合物与膦腈化合物的反应,进行含活性氢的化合物的脱质子化反应,其后,三异丁基铝发生作用,经脱质子化的含活性氢的化合物在铝上移动,从而成为催化活性种。
现有技术文献
专利文献
专利文献1:美国专利第5235114号说明书
专利文献2:日本特开平4-59825号公报
专利文献3:日本特许第3497054号说明书(日本特开平10-77289号公报)
专利文献4:日本特许第3905638号说明书(日本特开平11-106500号公报)
专利文献5:日本特许第5663856号说明书(日本特开2010-150514号公报)
专利文献6:日本特开2011-122134号公报
非专利文献
非专利文献1:Polymer Chemistry、2012、3、1189.
技术实现要素:
发明要解决的问题
专利文献1、2所述的方法中,所得聚环氧烷的分子量分布较宽,作为环氧烷存在难以应用于环氧乙烷等的课题。
专利文献3中提出的方法存在环氧烷的转化率低至9~19%的课题。此外,所得聚环氧烷是分子量为17000000~160000000g/mol的高分子量体与分子量为1700~2300g/mol的低分子量体的混合物,作为聚环氧烷没有特殊的特征。
专利文献4~6所述的方法中,所得聚环氧烷的不饱和度依然较高,寻求进一步降低。此外,若降低聚合温度,则产生催化活性降低的课题,反之,若提高聚合温度,则容易产生在产物中的杂质量的指标即不饱和度变高的课题。
非专利文献1中提出的方法需要大量使用昂贵的膦腈化合物,作为在工业上有效制造聚环氧烷的方法存在课题。此外,作为所得聚环氧烷也没有特殊的特征。
若聚环氧烷的不饱和度变高,则制成聚氨酯树脂时的交联密度降低、储能模量降低,因此存在滞后损耗、压缩永久形变等物性降低的课题。此外,若聚环氧烷的分子量分布变宽,则存在制成聚氨酯树脂时的成形性变差的课题。此外,若聚环氧烷中的低分子量成分变多,则制成聚氨酯树脂时的交联密度降低、储能模量降低,因此存在滞后损耗、压缩永久形变等物性降低的课题。
因而,寻求即使为低温的制造条件也能够高效地制造聚环氧烷的环氧烷聚合催化剂和使用其制造聚环氧烷的方法,所述聚环氧烷使用的昂贵膦腈化合物的量少、分子量高、不饱和度低且显示出狭窄的分子量分布。
用于解决问题的方案
本发明人等为了解决上述课题而进行了深入研究,结果发现:包含路易斯酸和特定膦腈盐的环氧烷聚合催化剂即使在低温的制造条件下也能够高效地制造分子量高、不饱和度低且分子量分布狭窄的聚环氧烷,从而完成了本发明。
即,本发明涉及以下示出的环氧烷聚合催化剂和使用其制造聚环氧烷的方法。
[1]一种环氧烷聚合催化剂,其包含路易斯酸和通式(1)所示的膦腈盐。
(上述通式(1)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者碳酸氢根阴离子。Y表示碳原子或磷原子,a在Y为碳原子时是2、在Y为磷原子时是3。)。
[2]根据上述[1]所述的环氧烷聚合催化剂,其特征在于,通式(1)所示的膦腈盐与路易斯酸的比例为[膦腈盐]:[路易斯酸]=1:0.002~500(摩尔比)。
[3]根据上述[1]或[2]所述的环氧烷聚合催化剂,其特征在于,路易斯酸为选自由铝化合物、锌化合物和硼化合物组成的组中的至少一种化合物。
[4]根据上述[1]或[2]所述的环氧烷聚合催化剂,其特征在于,路易斯酸为选自由有机铝、铝氧烷和有机锌组成的组中的至少一种化合物。
[5]根据上述[1]~[4]中任一项所述的环氧烷聚合催化剂,其包含通式(1)所示的膦腈盐、路易斯酸和含活性氢的化合物。
[上述通式(1)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者碳酸氢根阴离子。Y表示碳原子或磷原子,a在Y为碳原子时是2、在Y为磷原子时是3。]。
[6]根据上述[5]所述的环氧烷聚合催化剂,其特征在于,含活性氢的化合物为选自由水、羟基化合物、胺化合物、羧酸化合物、硫醇化合物和具有羟基的聚醚多元醇组成的组中的至少1种。
[7]一种上述[5]或[6]所述的环氧烷聚合催化剂的制造方法,其特征在于,将通式(1)所示的膦腈盐与含活性氢的化合物混合后,再将它们与路易斯酸混合。
[8]一种聚环氧烷的制造方法,其特征在于,在上述[1]~[5]中任一项所述的环氧烷聚合催化剂的存在下,进行环氧烷的开环聚合。
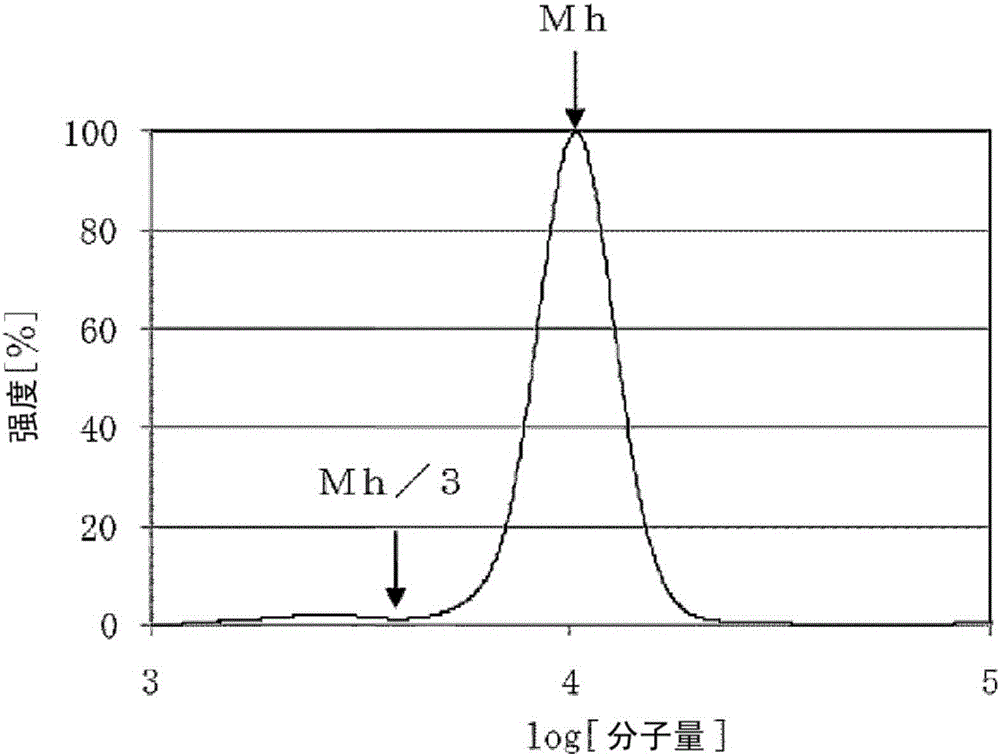
[9]一种聚环氧烷,其满足下述i)~iv)中的全部条件。
i)不饱和度为0.020meq/g以下
ii)Mw/Mn为1.10以下
iii)Mh/f为1000以上
iv)Mh/3以下的分子量的面积比率为2.0%以下
(其中,将以聚苯乙烯作为标准物质由凝胶渗透色谱测定求出的数均分子量记作Mn,将重均分子量记作Mw,将最高峰的分子量记作Mh,将聚环氧烷的官能团数量记作f。)
[10]根据上述[9]所述的聚环氧烷,其特征在于,由通过JIS K-1557记载的方法算出的聚环氧烷的羟基值和其官能团数量算出的分子量为1000~50000g/mol的范围。
[11]一种上述[9]或[10]所述的聚环氧烷的制造方法,其特征在于,在包含膦腈化合物和路易斯酸的环氧烷聚合催化剂的存在下,以含活性氢的化合物作为引发剂,进行环氧烷的开环聚合,并且,相对于前述含活性氢的化合物中的活性氢1摩尔,前述膦腈化合物的用量为0.001~0.1摩尔的范围。
[12]根据上述[11]所述的聚环氧烷的制造方法,其特征在于,膦腈化合物为通式(1)所示的膦腈盐。
(上述通式(1)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者碳酸氢根阴离子。Y表示碳原子或磷原子,a在Y为碳原子时是2、在Y为磷原子时是3。)。
[13]根据上述[11]或[12]所述的聚环氧烷的制造方法,其特征在于,路易斯酸为选自由铝化合物、锌化合物和硼化合物组成的组中的至少一种化合物。
[14]根据上述[11]或[12]所述的聚环氧烷的制造方法,其特征在于,路易斯酸为选自由有机铝、铝氧烷和有机锌组成的组中的至少一种化合物。
[15]根据上述[11]~[14]中任一项所述的聚环氧烷的制造方法,其特征在于,膦腈化合物与路易斯酸的比例为[膦腈化合物]:[路易斯酸]=1:0.002~500(摩尔比)。
发明的效果
根据本发明,能够获得由副反应导致的杂质生成量少、低温下的催化活性高的环氧烷聚合催化剂。此外,通过使用本发明的环氧烷聚合催化剂将环氧烷进行聚合,能够制造分子量高、不饱和度低且分子量分布狭窄的聚环氧烷。
此外,使用分子量高、不饱和度低、分子量分布狭窄、低分子量成分少的本发明的聚环氧烷而得到的聚氨酯树脂的储能模量提高,因此可期待滞后损耗、压缩永久形变等物性的提高。
附图说明
图1是实施例中的聚环氧烷的低分子量成分的面积比率的计算方法的示意图。
图2是实施例中的聚环氧烷的低分子量成分的面积比率的计算方法的示意图(放大图)。
具体实施方式
本发明的环氧烷聚合催化剂包含路易斯酸和通式(1)所示的膦腈盐。
[上述通式(1)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者碳酸氢根阴离子。Y表示碳原子或磷原子,a在Y为碳原子时是2、在Y为磷原子时是3。]。
此处,作为碳原子数1~20的烃基,可列举出例如甲基、乙基、乙烯基、正丙基、异丙基、环丙基、烯丙基、正丁基、异丁基、叔丁基、环丁基、正戊基、新戊基、环戊基、正己基、环己基、苯基、庚基、环庚基、辛基、环辛基、壬基、环壬基、癸基、环癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基等。
作为R1与R2相互键合而形成环结构的情况,可列举出例如吡咯烷基、吡咯基、哌啶基、吲哚基、异吲哚基等。
作为R1彼此相互键合或R2彼此相互键合而形成的环结构,可列举出例如一个取代基成为亚乙基、亚丙基、亚丁基等亚烷基且与另一个取代基相互键合而成的环结构。
并且,这些之中,作为R1和R2,由于形成催化活性优异的环氧烷聚合催化剂,因此,特别优选为甲基、乙基、异丙基。
上述膦腈盐中的X-为羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者碳酸氢根阴离子。
作为碳原子数1~4的烷氧基阴离子,可列举出例如甲氧基阴离子、乙氧基阴离子、正丙氧基阴离子、异丙氧基阴离子、正丁氧基阴离子、异丁氧基阴离子、叔丁氧基阴离子等。
作为碳原子数2~5的烷基羧基阴离子,可列举出例如乙酰氧基阴离子、乙基羧基阴离子、正丙基羧基阴离子、异丙基羧基阴离子、正丁基羧基阴离子、异丁基羧基阴离子、叔丁基羧基阴离子等。
这些之中,作为X-,由于形成催化活性优异的环氧烷聚合催化剂,因此,特别优选为羟基阴离子、碳酸氢根阴离子。
本发明中,Y为碳原子时,膦腈盐用下述通式(2)表示。
[上述通式(2)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者酸氢根阴离子。]
此外,本发明中,Y为磷原子时,膦腈盐用下述通式(3)表示。
[上述通式(3)中,R1和R2各自独立地表示氢原子或碳原子数1~20的烃基。此处,可以是R1与R2相互键合而形成环结构,也可以是R1彼此相互键合或R2彼此相互键合而形成环结构。X-表示羟基阴离子、碳原子数1~4的烷氧基阴离子、羧基阴离子、碳原子数2~5的烷基羧基阴离子、或者酸氢根阴离子。]
本发明中,作为膦腈盐,具体而言,可例示出四(1,1,3,3-四甲基胍基)氢氧化鏻、四(1,1,3,3-四乙基胍基)氢氧化鏻、四(1,1,3,3-四正丙基胍基)氢氧化鏻、四(1,1,3,3-四异丙基胍基)氢氧化鏻、四(1,1,3,3-四正丁基胍基)氢氧化鏻、四(1,1,3,3-四苯基胍基)氢氧化鏻、四(1,1,3,3-四苄基胍基)氢氧化鏻、四(1,3-二甲基咪唑烷-2-亚氨基)氢氧化鏻、四(1,3-二甲基咪唑烷-2-亚氨基)氢氧化鏻、四(1,1,3,3-四甲基胍基)碳酸氢鏻、四(1,1,3,3-四乙基胍基)碳酸氢鏻、四(1,1,3,3-四正丙基胍基)碳酸氢鏻、四(1,1,3,3-四异丙基胍基)碳酸氢鏻、四(1,1,3,3-四正丁基胍基)碳酸氢鏻、四(1,1,3,3-四苯基胍基)碳酸氢鏻、四(1,1,3,3-四苄基胍基)碳酸氢鏻、四(1,3-二甲基咪唑烷-2-亚氨基)碳酸氢鏻、四(1,3-二甲基咪唑烷-2-亚氨基)碳酸氢鏻等膦腈盐。
此外,可例示出四[三(二甲基氨基)膦烯氨基]氢氧化鏻、四[三(二乙基氨基)膦烯氨基]氢氧化鏻、四[三(二正丙基氨基)膦烯氨基]氢氧化鏻、四[三(二异丙基氨基)膦烯氨基]氢氧化鏻、四[三(二正丁基氨基)膦烯氨基]氢氧化鏻、四[三(二苯基氨基)膦烯氨基]氢氧化鏻、四[三(1,3-二甲基咪唑烷-2-亚氨基)膦烯氨基]氢氧化鏻、四[三(二甲基氨基)膦烯氨基]碳酸氢鏻、四[三(二乙基氨基)膦烯氨基]碳酸氢鏻、四[三(二正丙基氨基)膦烯氨基]碳酸氢鏻、四[三(二异丙基氨基)膦烯氨基]碳酸氢鏻、四[三(二正丁基氨基)膦烯氨基]碳酸氢鏻、四[三(二苯基氨基)膦烯氨基]碳酸氢鏻、四[三(1,3-二甲基咪唑烷-2-亚氨基)膦烯氨基]碳酸氢鏻等膦腈盐。
这些之中,由于形成催化性能优异的聚环氧烷制造催化剂,因此,特别优选为四(1,1,3,3-四甲基胍基)氢氧化膦腈、四(1,1,3,3-四甲基胍基)碳酸氢膦腈、四[三(二甲基氨基)膦烯氨基]氢氧化鏻。
本发明中,作为路易斯酸,可列举出例如铝化合物、锌化合物、硼化合物等。
作为铝化合物,可列举出例如有机铝、无机铝、铝氧烷等。
作为有机铝,可列举出例如三甲基铝、三乙基铝、三异丁基铝、三正己基铝、三乙氧基铝、三异丙氧基铝、三异丁氧基铝、三苯基铝、二苯基单异丁基铝、单苯基二异丁基铝等。
作为无机铝,可列举出氯化铝、氢氧化铝、氧化铝等。
作为铝氧烷,可列举出例如下式所示的化合物。
(-AlR3-O-)n
(式中,R3表示碳原子数1~20的烃基。n表示2以上的整数。)
此处,作为碳原子数1~20的烃基,可列举出例如甲基、乙基、乙烯基、正丙基、异丙基、环丙基、烯丙基、正丁基、异丁基、叔丁基、环丁基、正戊基、新戊基、环戊基、正己基、环己基、苯基、庚基、环庚基、辛基、环辛基、壬基、环壬基、癸基、环癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基等。这些之中,作为R3,由于形成容易获取且显示出高环氧烷转化率的环氧烷聚合催化剂,因此优选为甲基、异丁基。
作为铝氧烷的具体例,可列举出甲基铝氧烷、异丁基铝氧烷、甲基异丁基铝氧烷等。此外,作为铝氧烷的市售品,可列举出例如(商品名)MMAO-3A(Tosoh Finechem Corporation制)、(商品名)TMAO-340系列(Tosoh Finechem Corporation制)、(商品名)TMAO-200系列(Tosoh Finechem Corporation制)、(商品名)PBAO(Tosoh Finechem Corporation制)、(商品名)Solid-MAO(Tosoh Finechem Corporation制)等。
作为锌化合物,可列举出例如二甲基锌、二乙基锌、二苯基锌等有机锌;氯化锌、氧化锌等无机锌。
作为硼化合物,可列举出三乙基硼烷、三甲氧基硼烷、三乙氧基硼烷、三异丙氧基硼烷、三苯基硼烷、三(五氟苯基)硼烷、三氟硼烷等。
并且,这些之中,由于形成催化性能优异的环氧烷聚合催化剂,因此优选为有机铝、铝氧烷、有机锌,特别优选为有机铝。
此外,这些之中,由于形成容易获取且催化活性优异的环氧烷聚合催化剂,因此,特别优选为三异丁基铝、三异丙氧基铝、二乙基锌。
本发明的环氧烷聚合催化剂中的膦腈盐与路易斯酸的比例只要表现出作为环氧烷聚合催化剂的作用就是任意的,没有特别限定。这些之中,尤其是形成催化活性优异、环氧烷的转化率高的聚合催化剂,因此优选为[膦腈盐]:[路易斯酸]=1:0.002~500(摩尔比)。
作为本发明的环氧烷聚合催化剂,由于能够高效地制造分子量更高、不饱和度更低、且显示出狭窄分子量分布的聚环氧烷,形成催化活性优异的聚合催化剂,因此,进一步特别优选使用含活性氢的化合物。作为此时的含活性氢的化合物,可列举出例如水、羟基化合物、胺化合物、羧酸化合物、硫醇化合物、具有羟基的聚醚多元醇等。
作为羟基化合物,可列举出例如乙二醇、二乙二醇、丙二醇、二丙二醇、1,3-丙二醇、1,3-丁二醇、1,4-丁二醇、1,6-己二醇、甘油、三羟甲基丙烷、己三醇、季戊四醇、二甘油、山梨糖醇、蔗糖、葡萄糖、2-萘酚、双酚等。
作为胺化合物,可列举出例如乙二胺、N,N’-二甲基乙二胺、哌啶、哌嗪等。
作为羧酸化合物,可列举出例如苯甲酸、己二酸等。
作为硫醇化合物,可列举出例如乙二硫醇、丁二硫醇等。
作为具有羟基的聚醚多元醇,可列举出例如分子量为200~3000的聚醚多元醇等。
并且,这些含活性氢的化合物可以单独使用,也可以混合多种使用。
此外,使用含活性氢的化合物时,由于能够形成可高效地制造分子量更高、不饱和度更低、且显示出狭窄分子量分布的聚环氧烷且催化活性优异的聚合催化剂,因此,相对于含活性氢的化合物中的活性氢1摩尔,上述膦腈盐优选为0.001~10摩尔、特别优选为0.001~5摩尔。此外,相对于含活性氢的化合物中的活性氢1摩尔,上述路易斯酸优选为0.001~10摩尔、特别优选为0.001~5摩尔。
本发明中,环氧烷聚合催化剂的制备方法只要能够形成本发明的聚环氧烷,则可以使用任何方法,没有特别限定。
可列举出例如将膦腈盐与路易斯酸进行混合的方法。此时,作为溶剂,可以使用例如苯、甲苯、二甲苯、环己烷、1,2-二氯乙烷、氯苯、二氯苯、1,4-二噁烷、1,2-二甲氧基乙烷等。
此外,在包含膦腈盐和路易斯酸的环氧烷聚合催化剂的存在下,以含活性氢的化合物作为引发剂来进行环氧烷的开环聚合时,可以使用将膦腈盐、路易斯酸和含活性氢的化合物同时混合的方法;向这些之中的1种成分中混合其它的2种成分的方法;向这些之中的2种成分中混合其它的1种成分的方法等任意方法。
这些之中,由于能够制备可高效地制造分子量更高、不饱和度更低、且显示出狭窄分子量分布的聚环氧烷且催化活性优异的聚合催化剂,因此优选的是,将上述膦腈盐与含活性氢的化合物混合后,再将它们与路易斯酸混合,从而制备环氧烷聚合催化剂。此时,也可以进行加热/减压处理等,作为加热处理的温度,可列举出例如50~150℃、优选为70~130℃,此外,作为减压处理时的压力,可列举出例如50kPa以下、优选为20kPa以下。
本发明的环氧烷聚合催化剂的催化活性优异,因此对于聚环氧烷的制造而言是有用的,可以在本发明的环氧烷聚合催化剂的存在下进行环氧烷的开环聚合。
作为此时的环氧烷,没有特别限定,可列举出例如碳原子数2~20的环氧烷。具体而言,可例示出环氧乙烷、环氧丙烷、1,2-环氧丁烷、2,3-环氧丁烷、异环氧丁烷、一氧化丁二烯、环氧戊烷、环氧苯乙烯、环氧环己烯等。
这些之中,由于容易获取环氧烷且所得聚环氧烷的工业价值高,因此优选为环氧乙烷、环氧丙烷。环氧烷可以单独使用,也可以混合2种以上使用。混合2种以上使用时,例如可以使第一环氧烷反应后再与第二环氧烷反应,还可以使2种以上的环氧烷同时反应。
本发明的聚环氧烷的制造方法中,聚合压力可以为0.05~1.0MPa的范围、优选为0.1~0.6MPa的范围。本发明的聚环氧烷的制造中,聚合温度可以为0~130℃的范围、优选为10~110℃的范围。
本发明的聚环氧烷的制造方法中,聚合可以在溶剂中进行,或者在无溶剂的条件下进行。作为使用的溶剂,没有特别限定,可列举出例如苯、甲苯、二甲苯、环己烷、1,2-二氯乙烷、氯苯、二氯苯、1,4-二噁烷、1,2-二甲氧基乙烷等。
本发明的聚环氧烷的制造方法中,由于成为有效的聚环氧烷的制造方法,因此,作为催化活性,优选显示出100g/mol·min以上的催化活性、特别优选显示出200g/mol·min以上的催化活性。
关于通过本发明的制造方法而得到的聚环氧烷,作为由通过JIS K-1557记载的方法算出的聚环氧烷的羟基值和其官能团数量算出的分子量,优选为1000~50000g/mol、特别优选为3000~30000g/mol。
此外,不饱和度优选为0.05meq/g以下、特别优选为0.03meq/g以下。进而,由聚环氧烷的数均分子量(Mn)、重均分子量(Mw)算出的聚环氧烷的分子量分布(Mw/Mn)优选为1.3以下、特别优选为1.1以下。
接着,针对满足本发明的下述i)~iv)中的全部条件的聚环氧烷进行说明。
i)不饱和度为0.020meq/g以下
ii)Mw/Mn为1.10以下
iii)Mh/f为1000以上
iv)Mh/3以下的分子量的面积比率为2.0%以下。
[其中,将以聚苯乙烯作为标准物质由凝胶渗透色谱测定求出的数均分子量记作“Mn”、将重均分子量记作“Mw”、将最高峰的分子量记作“Mh”、将聚环氧烷的官能团数量记作“f”。]
本发明的聚环氧烷的不饱和度为0.020meq/g以下、优选为0.010meq/g以下。若不饱和度大于0.020meq/g,则制成聚氨酯树脂时的储能模量降低,滞后损耗、压缩永久形变等物性降低,故不优选。
本发明的聚环氧烷的Mw/Mn为1.10以下、优选为1.08以下。若Mw/Mn大于1.10,则制成聚氨酯树脂时的储能模量降低、成形性变差,故不优选。
本发明的聚环氧烷的Mh/f为1000以上、优选为1500以上。若Mh/f小于1000,则制成聚氨酯树脂时的柔软性等物性变差,故不优选。
本发明的聚环氧烷的Mh/3以下的分子量的面积比率为2.0%以下、优选为1.0%以下。若Mh/3以下的分子量的面积比率大于2.0%,则制成聚氨酯树脂时的储能模量降低,滞后损耗、压缩永久形变等物性降低,故不优选。
关于本发明的聚环氧烷,作为由通过上述JIS K-1557记载的方法算出的聚环氧烷的羟基值和其官能团数量算出的分子量,优选为1000~50000g/mol、特别优选为3000~30000g/mol。
本发明的聚环氧烷例如通过在包含膦腈化合物和路易斯酸的环氧烷聚合催化剂的存在下,以含活性氢的化合物作为引发剂来进行环氧烷的开环聚合,并且,将前述膦腈化合物相对于前述含活性氢的化合物中的活性氢1摩尔的用量设为0.001~0.1摩尔的范围,从而能够简便地制造。
此处,作为膦腈化合物,具体而言,可例示出四(1,1,3,3-四甲基胍基)氢氧化鏻、四(1,1,3,3-四乙基胍基)氢氧化鏻、四(1,1,3,3-四正丙基胍基)氢氧化鏻、四(1,1,3,3-四异丙基胍基)氢氧化鏻、四(1,1,3,3-四正丁基胍基)氢氧化鏻、四(1,1,3,3-四苯基胍基)氢氧化鏻、四(1,1,3,3-四苄基胍基)氢氧化鏻、四(1,3-二甲基咪唑烷-2-亚氨基)氢氧化鏻、四(1,3-二甲基咪唑烷-2-亚氨基)氢氧化鏻、四(1,1,3,3-四甲基胍基)碳酸氢鏻、四(1,1,3,3-四乙基胍基)碳酸氢鏻、四(1,1,3,3-四正丙基胍基)碳酸氢鏻、四(1,1,3,3-四异丙基胍基)碳酸氢鏻、四(1,1,3,3-四正丁基胍基)碳酸氢鏻、四(1,1,3,3-四苯基胍基)碳酸氢鏻、四(1,1,3,3-四苄基胍基)碳酸氢鏻、四(1,3-二甲基咪唑烷-2-亚氨基)碳酸氢鏻、四(1,3-二甲基咪唑烷-2-亚氨基)碳酸氢鏻等膦腈盐。
此外,可例示出四[三(二甲基氨基)膦烯氨基]氢氧化鏻、四[三(二乙基氨基)膦烯氨基]氢氧化鏻、四[三(二正丙基氨基)膦烯氨基]氢氧化鏻、四[三(二异丙基氨基)膦烯氨基]氢氧化鏻、四[三(二正丁基氨基)膦烯氨基]氢氧化鏻、四[三(二苯基氨基)膦烯氨基]氢氧化鏻、四[三(1,3-二甲基咪唑烷-2-亚氨基)膦烯氨基]氢氧化鏻、四[三(二甲基氨基)膦烯氨基]碳酸氢鏻、四[三(二乙基氨基)膦烯氨基]碳酸氢鏻、四[三(二正丙基氨基)膦烯氨基]碳酸氢鏻、四[三(二异丙基氨基)膦烯氨基]碳酸氢鏻、四[三(二正丁基氨基)膦烯氨基]碳酸氢鏻、四[三(二苯基氨基)膦烯氨基]碳酸氢鏻、四[三(1,3-二甲基咪唑烷-2-亚氨基)膦烯氨基]碳酸氢鏻等膦腈盐。
此外,可例示出1-叔丁基-4,4,4-三(二甲基氨基)-2,2-双(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-联二膦腈。
此处,作为膦腈化合物,优选为上述通式(1)所示的膦腈盐。
本发明的聚环氧烷由于分子量高、不饱和度低、分子量分布狭窄、低分子量成分少,因此,可期待使用其得到的聚氨酯树脂的储能模量提高,滞后损耗、压缩永久形变等物性提高。
实施例
以下,通过实施例来说明本发明,但本实施例完全不限定本发明。
首先,针对催化活性的计算方法、聚环氧烷的分析方法进行说明。
(1)催化活性(单位:g/mol·min)
将已反应的环氧烷量记作a(单位:g),将使用的膦腈盐量记作b(单位:mol),将聚合所需的时间记作c(单位:min),通过下式来算出催化活性。
催化活性=a/(b×c)。
(2)聚环氧烷的分子量(单位:g/mol)
通过JIS K-1557记载的方法,测定聚环氧烷的羟基值d(单位:mgKOH/g)。将所得聚环氧烷的官能团数量记作e,通过下式来算出聚环氧烷的分子量。
分子量=(56100/d)×e。
此外,使用凝胶渗透色谱(GPC)(东曹株式会社制、HLC8020),以四氢呋喃作为溶剂,在40℃下进行测定,作为标准物质而使用聚苯乙烯,算出聚环氧烷的数均分子量(Mn)、重均分子量(Mw)、最高峰的分子量(Mh)。
(3)聚环氧烷的低分子量成分的面积比率(单位:%)
计算通过上述方法(2)算出的Mh除以3而得到的分子量(Mh/3)以下、即低分子量成分的面积比率。
(4)聚环氧烷的不饱和度(单位:meq/g)
通过JIS K-1557记载的方法,算出聚环氧烷的不饱和度。
(5)聚环氧烷的分子量分布(单位:无)
根据通过上述方法(2)算出的数均分子量(Mn)、重均分子量(Mw),算出该聚环氧烷的分子量分布(Mw/Mn)。
合成例1(膦腈盐A的合成)
将附带搅拌叶片的2升四颈烧瓶中制成氮气气氛,添加五氯化磷96g(0.46mol)、脱水甲苯800ml,以20℃进行搅拌。在维持搅拌的条件下,耗费3小时滴加1,1,3,3-四甲基胍345g(2.99mol)后,升温至100℃,进一步耗费1小时滴加1,1,3,3-四甲基胍107g(0.92mol)。将所得白色的浆料溶液以100℃搅拌14小时后,冷却至80℃,添加离子交换水250ml,搅拌30分钟。停止搅拌时,浆料全部溶解而得到二相溶液。进行所得二相溶液的油水分离,回收水相。向所得水相中添加二氯甲烷100ml,进行油水分离,回收二氯甲烷相。将所得二氯甲烷溶液用离子交换水100ml进行清洗。
将所得二氯甲烷溶液转移至附带搅拌叶片的2升四颈烧瓶中,添加2-丙醇900g后,在常压下将温度提升至80~100℃,去除二氯甲烷。一边搅拌所得2-丙醇溶液,一边将内部温度放冷至60℃后,添加85重量%的氢氧化钾31g(0.47mol、相对于亚氨基膦腈盐为1.1mol当量),以60℃反应2小时。将温度冷却至25℃,通过过滤而去除所析出的副产盐,从而以25重量%的浓度、92%的收率得到作为目标的亚氨基膦腈盐A[上述通式(1)中的R1为甲基、R2为甲基、X-为羟基阴离子、Y为碳原子、a相当于2的膦腈盐]的2-丙醇溶液860g。
合成例2(膦腈盐B的合成)
将附带磁性转子的100ml舒伦克瓶制成氮气气氛,添加四[三(二甲基氨基)膦烯氨基]氯化磷5.7g(7.4mmol、Aldrich公司制)、2-丙醇16ml,以25℃进行搅拌而使其溶解。在维持搅拌的条件下,添加将85重量%的氢氧化钾0.53g〔8.1mmol、相对于四[三(二甲基氨基)膦烯氨基]氯化鏻为1.1mol当量〕溶解于2-丙醇而得到的溶液。以25℃搅拌5小时后,通过过滤而去除所析出的副产盐,从而以17重量%的浓度、98%的收率得到作为目标的膦腈盐B[上述通式(1)中的R1为甲基、R2为甲基、X-为羟基阴离子、Y为磷原子、a相当于3的膦腈盐]的2-丙醇溶液32.7g。
实施例1
向附带搅拌叶片的0.2升高压釜中添加通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液10g(5mmol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下去除2-丙醇。其后,添加三异丁基铝的1.0mol/l的甲苯溶液10ml(10mmol)并混合,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成20℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷108g,一边在内部温度为18~22℃的范围内发生反应。在0.5kPa的减压下进行残留环氧丙烷和甲苯的去除,从而得到无色无臭的聚环氧烷106g。催化活性为350g/mol·min、所得聚环氧烷的分子量为20000g/mol、不饱和度为0.018meq/g、分子量分布为1.08。将结果示于表1。
实施例2
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相对于活性氢1mol为0.010mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为840g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.017meq/g、分子量分布为1.07。将结果示于表1。
实施例3
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.27g(0.14mmol、相对于活性氢1mol为0.0025mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.27ml(0.27mmol、相对于活性氢1mol为0.005mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷126g。催化活性为1100g/mol·min、所得聚环氧烷的分子量为6800g/mol、不饱和度为0.016meq/g、分子量分布为1.06。将结果示于表1。
实施例4
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相对于活性氢1mol为0.010mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成50℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为48~52℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为48~52℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷125g。催化活性为550g/mol·min、所得聚环氧烷的分子量为6800g/mol、不饱和度为0.014meq/g、分子量分布为1.05。将结果示于表1。
实施例5
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)6.0g(6.0mmol、活性氢量为18mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.36g(0.18mmol、相对于活性氢1mol为0.010mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.36ml(0.36mmol、相对于活性氢1mol为0.020mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷112g。催化活性为980g/mol·min、所得聚环氧烷的分子量为18000g/mol、不饱和度为0.022meq/g、分子量分布为1.08。将结果示于表1。
实施例6
使用二乙基锌的1.0mol/l的己烷溶液0.54ml(0.54mmol、相对于活性氢1mol为0.010mol)来代替三异丁基铝的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相对于活性氢1mol为0.010mol),除此之外,通过与实施例2相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。得到无色无臭的聚环氧烷127g。催化活性为750g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.016meq/g、分子量分布为1.06。将结果示于表1。
[表1]
比较例1
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)6.0g(6.0mmol、活性氢量为18mmol)、以及作为膦腈P4碱的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-双(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-联二膦腈的1.0mol/l的己烷溶液18ml(18mmol、相对于活性氢1mol为1.0mol)。将高压釜内制成氮气气氛后,添加三异丁基铝的2.0mol/l的甲苯溶液18ml(36mmol、相对于活性氢1mol为2.0mol)并混合,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成20℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷37g,一边在内部温度为18~22℃的范围内发生反应。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷42g。催化活性为11g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.016meq/g、分子量分布为1.36。将结果示于表2。
比较例2
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷,一边在内部温度为68~72℃的范围内发生反应。其结果,在供给36g环氧丙烷的时刻,反应终止。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷36g。催化活性为46g/mol·min、所得聚环氧烷的分子量为2000g/mol、不饱和度为0.013meq/g、分子量分布为1.05。将结果示于表2。
[表2]
实施例7
向附带搅拌叶片的0.2升高压釜中添加通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液10g(5mmol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下去除2-丙醇。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液10ml(铝原子10mmol)并混合,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成35℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷108g,一边在内部温度为33~37℃的范围内发生反应。在0.5kPa的减压下进行残留环氧丙烷和甲苯的去除,从而得到无色无臭的聚环氧烷106g。环氧烷的转化率为98%、所得聚环氧烷的分子量为19000g/mol、不饱和度为0.016meq/g、分子量分布为1.07。将结果示于表3。
实施例8
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.54ml(铝原子为0.54mmol、相对于活性氢1mol为0.010mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。环氧烷的转化率为99%、所得聚环氧烷的分子量为7000g/mol、不饱和度为0.016meq/g、分子量分布为1.06。将结果示于表3。
实施例9
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.27g(0.14mmol、相对于活性氢1mol为0.0025mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.27ml(铝原子为0.27mmol、相对于活性氢1mol为0.005mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷126g。环氧烷的转化率为98%、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.014meq/g、分子量分布为1.05。将结果示于表3。
实施例10
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.54ml(铝原子为0.54mmol、相对于活性氢1mol为0.010mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成50℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为48~52℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为48~52℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷125g。环氧烷的转化率为97%、所得聚环氧烷的分子量为6700g/mol、不饱和度为0.012meq/g、分子量分布为1.04。将结果示于表3。
实施例11
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)6.0g(6.0mmol、活性氢量为18mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.36g(0.18mmol、相对于活性氢1mol为0.010mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.36ml(铝原子为0.36mmol、相对于活性氢1mol为0.020mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷112g。环氧烷的转化率为96%、所得聚环氧烷的分子量为18000g/mol、不饱和度为0.021meq/g、分子量分布为1.08。将结果示于表3。
[表3]
比较例3
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)6.0g(6.0mmol、活性氢量为18mmol)、以及具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液36ml(铝原子36mmol、相对于活性氢1mol为2.0mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成35℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷,一边在内部温度为33~37℃的范围内发生反应。其结果,在供给12g环氧丙烷的时刻,反应终止。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷9.0g。环氧烷的转化率为25%、所得聚环氧烷的分子量为1500g/mol、不饱和度为0.012meq/g、分子量分布为1.42。将结果示于表4。
比较例4
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)18g(18mmol、活性氢量为54mmol)、以及通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷,一边在内部温度为68~72℃的范围内发生反应。其结果,在供给36g环氧丙烷的时刻,反应终止。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷36g。环氧烷的转化率为50%、所得聚环氧烷的分子量为2000g/mol、不饱和度为0.013meq/g、分子量分布为1.05。将结果示于表4。
[表4]
实施例12
向附带搅拌叶片的0.2升高压釜中添加通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液11g(2.5mmol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下去除2-丙醇。其后,添加三异丁基铝的1.0mol/l的甲苯溶液7.5ml(7.5mmol)并混合,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成20℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷108g,一边在内部温度为68~72℃的范围内发生反应。在0.5kPa的减压下进行残留环氧丙烷和甲苯的去除,从而得到无色无臭的聚环氧烷106g。催化活性为400g/mol·min、所得聚环氧烷的分子量为20000g/mol、不饱和度为0.008meq/g、分子量分布为1.08。将结果示于表5。
实施例13
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]18g(18mmol、活性氢量为54mmol)、以及通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液2.0g(0.45mmol、相对于活性氢1mol为0.008mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为108~112℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为1040g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.007meq/g、分子量分布为1.07。将结果示于表5。
实施例14
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]18g(18mmol、活性氢量为54mmol)、以及通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液1.0g(0.23mmol、相对于活性氢1mol为0.004mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.68ml(0.68mmol、相对于活性氢1mol为0.013mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为108~112℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷126g。催化活性为1300g/mol·min、所得聚环氧烷的分子量为6800g/mol、不饱和度为0.005meq/g、分子量分布为1.06。将结果示于表5。
实施例15
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]18g(18mmol、活性氢量为54mmol)、以及通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液1.2g(0.27mmol、相对于活性氢1mol为0.005mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相对于活性氢1mol为0.010mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为78~82℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为108~112℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷125g。催化活性为650g/mol·min、所得聚环氧烷的分子量为6800g/mol、不饱和度为0.007meq/g、分子量分布为1.05。将结果示于表5。
实施例16
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇(三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g)6.0g(6.0mmol、活性氢量为18mmol)、以及通过合成例2得到的膦腈盐B的25重量%2-丙醇溶液0.54g(0.18mmol、相对于活性氢1mol为0.010mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理。其后,添加三异丁基铝的1.0mol/l的甲苯溶液0.36ml(0.36mmol、相对于活性氢1mol为0.020mol),将内部温度制成80℃,进行0.5kPa的减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边在内部温度为68~72℃的范围内发生反应。接着,在0.5kPa的减压下进行残留环氧丙烷的去除,然后一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边在内部温度为108~112℃的范围内发生反应。在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷112g。催化活性为980g/mol·min、所得聚环氧烷的分子量为18000g/mol、不饱和度为0.008meq/g、分子量分布为1.08。将结果示于表5。
实施例17
使用三异丙氧基铝0.15g(0.75mmol、相对于活性氢1mol为0.025mol)来代替三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.008mol),除此之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷127g。催化活性为750g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.007meq/g、分子量分布为1.05。将结果示于表5。
[表5]
实施例18
使用三苯基铝1.0M溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol)来代替三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),除此之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷127g。催化活性为680g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.006meq/g、分子量分布为1.06。将结果示于表6。
实施例19
使用氯化铝0.18g(1.35mmol、相对于活性氢1mol为0.025mol)来代替三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),除此之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷126g。催化活性为650g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.006meq/g、分子量分布为1.07。将结果示于表6。
实施例20
使用纯异丁基铝氧烷1.0M己烷溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol)来代替三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),除此之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷107g。催化活性为350g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.006meq/g、分子量分布为1.06。将结果示于表6。
实施例21
除了将高压釜的内部温度从70℃变更为120℃之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷127g。催化活性为850g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.006meq/g、分子量分布为1.06。将结果示于表6。
实施例22
使用二乙基锌的1.0mol/l的己烷溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol)来代替三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),除此之外,通过与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷127g。催化活性为750g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.010meq/g、分子量分布为1.07。将结果示于表6。
[表6]
比较例5
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]6.0g(6.0mmol、活性氢量为18mmol)、以及作为膦腈P4碱的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-双(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-联二膦腈的1.0mol/l的己烷溶液18ml(18mmol、相对于活性氢1mol为1.0mol)。将高压釜内制成氮气气氛后,添加三异丁基铝的2.0mol/l的甲苯溶液18ml(36mmol、相对于活性氢1mol为2.0mol)并混合,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成20℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷37g,一边在内部温度为18~22℃的范围内发生反应。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷42g。催化活性为11g/mol·min、所得聚环氧烷的分子量为6900g/mol、不饱和度为0.016meq/g、分子量分布为1.36。将结果示于表7。
比较例6
除了将膦腈盐0.45mmol设为KOH 0.45mmol之外,利用与实施例13相同的方法,进行环氧烷聚合催化剂、聚环氧烷的制造。从而得到无色无臭的聚环氧烷36g。催化活性为61g/mol·min、所得聚环氧烷的分子量为2000g/mol、不饱和度为0.030meq/g、分子量分布为1.05。将结果示于表7。
比较例7
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]18g(18mmol、活性氢量为54mmol)、以及通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液2.0g(0.45mmol、相对于活性氢1mol为0.008mol)。将高压釜内制成氮气气氛后,将内部温度制成80℃,在0.5kPa的减压下进行脱水处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷,一边在内部温度为88~92℃的范围内发生反应。其结果,供给了110g环氧丙烷。在0.5kPa的减压下进行残留环氧丙烷的去除,从而得到无色无臭的聚环氧烷125g。催化活性为460g/mol·min、所得聚环氧烷的分子量为7000g/mol、不饱和度为0.024meq/g、分子量分布为1.06。将结果示于表3。
比较例8
向附带搅拌叶片的0.2升高压釜中添加作为含活性氢的化合物的具有3个羟基且分子量为1000的聚醚多元醇[三洋化成工业株式会社制、(商品名)SANNIX GP1000;羟基值为160mgKOH/g]18g(18mmol、活性氢量为54mmol)、三异丁基铝的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相对于活性氢1mol为0.025mol),将内部温度制成80℃,进行0.5kPa的减压处理。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,使环氧丙烷在反应压力0.3MPa以下、内部温度68~72℃的范围内发生反应。然而,在70℃的反应温度下,反应压力达到0.3MPa后,完全未观察到压力的降低。以70℃持续反应5小时后,在0.5kPa的减压下进行残留环氧丙烷的去除。回收10g作为原料的聚醚多元醇。三异丁基铝单独存在时,完全显示不出环氧丙烷的聚合活性。将结果示于表7。
[表7]
实施例23
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为600g/mol·min、所得聚环氧烷的不饱和度为0.006meq/g、分子量分布为1.06、Mh/f为3700g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
实施例24
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液0.54ml(0.54mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷126g。催化活性为600g/mol·min、所得聚环氧烷的不饱和度为0.005meq/g、分子量分布为1.05、Mh/f为3600g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
实施例25
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成70℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷125g。催化活性为400g/mol·min、所得聚环氧烷的不饱和度为0.005meq/g、分子量分布为1.05、Mh/f为3600g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
实施例26
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成120℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷128g。催化活性为1500g/mol·min、所得聚环氧烷的不饱和度为0.008meq/g、分子量分布为1.06、Mh/f为3700g/mol、Mh/3以下的低分子量成分的面积比率为0.2%。
实施例27
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)9g(活性氢量27mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.45g(0.23mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液0.68ml(0.68mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷117g。催化活性为500g/mol·min、所得聚环氧烷的不饱和度为0.008meq/g、分子量分布为1.06、Mh/f为7000g/mol、Mh/3以下的低分子量成分的面积比率为0.2%。
实施例28
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷109g。催化活性为500g/mol·min、所得聚环氧烷的不饱和度为0.008meq/g、分子量分布为1.06、Mh/f为7000g/mol、Mh/3以下的低分子量成分的面积比率为0.2%。
实施例29
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加异丙醇铝(Al(OiPr)3)的1.0mol/l己烷溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为600g/mol·min、所得聚环氧烷的不饱和度为0.005meq/g、分子量分布为1.08、Mh/f为3500g/mol、Mh/3以下的低分子量成分的面积比率为1.4%。
实施例30
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三苯基铝(AlPh3)的1.0mol/l二丁醚溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为600g/mol·min、所得聚环氧烷的不饱和度为0.006meq/g、分子量分布为1.07、Mh/f为3700g/mol、Mh/3以下的低分子量成分的面积比率为0.4%。
实施例31
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加具有甲基和异丁基的铝氧烷(Tosoh Finechem Corporation制、MMAO-3A)1.0mol/l己烷溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷85g。催化活性为250g/mol·min、所得聚环氧烷的不饱和度为0.005meq/g、分子量分布为1.05、Mh/f为2000g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
实施例32
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.90g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加二乙基锌(ZnEt2)的1.0mol/l己烷溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷85g。催化活性为250g/mol·min、所得聚环氧烷的不饱和度为0.005meq/g、分子量分布为1.05、Mh/f为2000g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
实施例33
将附带搅拌叶片的0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制SANNIX GP1000)18g(活性氢量为54mmol)、通过合成例2得到的膦腈盐B的17重量%2-丙醇溶液2.0g(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为600g/mol·min、所得聚环氧烷的不饱和度为0.006meq/g、分子量分布为1.06、Mh/f为3700g/mol、Mh/3以下的低分子量成分的面积比率为0.1%。
将上述结果合并示于表8、表9。
[表8]
[表9]
由表8、表9明确得那样,通过实施例23~例33得到的聚环氧烷满足下述i)~iv)中的全部条件。
i)不饱和度为0.020meq/g以下
ii)Mw/Mn为1.10以下
iii)Mh/f为1000以上
iv)Mh/3以下的分子量的面积比率为2.0%以下
(其中,将以聚苯乙烯作为标准物质由凝胶渗透色谱测定求出的数均分子量记作Mn,将重均分子量记作Mw、将最高峰的分子量记作Mh,将聚环氧烷的官能团数量记作f。)。
比较例9
将0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(18mmol)、通过合成例1得到的膦腈盐A的25重量%2-丙醇溶液0.54g(0.27mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷92g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。接着,将高压釜的内部温度制成110℃,一边以反应压力保持为0.25MPa以下的方式间歇地供给环氧乙烷18g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧乙烷的去除,从而得到无色无臭的聚环氧烷127g。催化活性为500g/mol·min、所得聚环氧烷的不饱和度为0.026meq/g、分子量分布为1.12、Mh/f为3300g/mol、Mh/3以下的低分子量成分的面积比率为3.7%。
比较例10
将0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(18mmol)、三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。
将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷10g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。从而得到无色无臭的聚环氧烷18g。催化活性为0g/mol·min、所得聚环氧烷为作为原料的聚醚多元醇(三洋化成工业株式会社制SANNIX GP1000)。
比较例11
将0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)18g(活性氢量为54mmol)、氢氧化钾(KOH)的50重量%水溶液50mg(0.45mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成90℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷36g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。从而得到无色无臭的聚环氧烷27g。催化活性为60g/mol·min、所得聚环氧烷的不饱和度为0.030meq/g、分子量分布为1.12、Mh/f为500g/mol、Mh/3以下的低分子量成分的面积比率为4.3%。
比较例12
将0.2升高压釜制成氮气气氛,添加聚醚多元醇(三洋化成工业株式会社制、SANNIX GP1000)6g(活性氢量为18mmol)、以及作为膦腈P4碱的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-双(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-联二膦腈的1.0mol/l的己烷溶液18ml(18mmol)。将内部温度制成80℃,在0.5kPa下进行减压处理。其后,添加三异丁基铝(TIBAL)的2.0mol/l甲苯溶液18ml(36mmol),将内部温度制成80℃,在0.5kPa下进行减压处理,从而得到环氧烷聚合催化剂。
在所得环氧烷聚合催化剂的存在下,将高压釜的内部温度制成20℃,一边以反应压力保持为0.3MPa以下的方式间歇地供给环氧丙烷37g,一边使其反应。反应结束后,在0.5kPa的减压下进行残留环氧丙烷的去除。从而得到无色无臭的聚环氧烷42g。催化活性为10g/mol·min、所得聚环氧烷的不饱和度为0.016meq/g、分子量分布为1.36、Mh/f为3,300g/mol、Mh/3以下的低分子量成分的面积比率为3.7%。
将比较例9~12的结果示于表10。
[表10]
应用例1
将通过实施例23得到的聚环氧烷100重量份、二苯基甲烷二异氰酸酯5.8份(商品名:C-1394)、三乙二胺0.05份(商品名:TEDA-L33)投入至デスポカップ中,并进行搅拌。搅拌结束后,使用旋转型流变仪(UBM公司制造的ソリキッドメーターMR-500),在氮气气氛下在25℃、频率10Hz的条件下测定粘弹性。自搅拌结束起50分钟后的储能模量(G’)为45,000Pa。
应用例2
将通过比较例9得到的聚环氧烷100重量份、二苯基甲烷二异氰酸酯5.8份(商品名:C-1394)、三乙二胺0.05份(商品名:TEDA-L33)投入至一次性杯子(diaposable cup)中,并进行搅拌。搅拌结束后,使用旋转型流变仪(UBM公司制造的ソリキッドメーターMR-500),在氮气气氛下在25℃、频率10Hz的条件下测定粘弹性。自搅拌结束起50分钟后的储能模量(G’)为15000Pa。
如上所示,参照特定的实施方式针对本发明进行了详细说明,但在不超脱本发明本质和范围的条件下能够施加各种变更、修正对于本领域技术人员而言是不言而喻的。
本发明中,通式(1)所示的膦腈盐(Y、R1、R2、X)与路易斯酸与根据需要而使用的含活性氢的化合物的适合组合例如如表11~18所示那样。
[表11]
[表12]
[表13]
[表14]
[表15]
[表16]
[表17]
[表18]
此外,本发明中,膦腈化合物与路易斯酸与根据需要使用的含活性氢的化合物的适合组合具体而言如表19~24所示那样。
[表19]
[表20]
[表21]
[表22]
[表23]
[表24]
应予说明,将2014年8月12日申请的日本特愿2014-164289号的说明书、权利要求书和摘要、2014年8月12日申请的日本特愿2014-164290号的说明书、权利要求书和摘要、2015年7月23日申请的日本特愿2015-145645号的说明书、权利要求书和摘要、以及2015年7月23日申请的日本特愿2015-145646号的说明书、权利要求书、附图和摘要的全部内容援引至此,作为本发明说明书的公开内容。
产业上的可利用性
通过使用本发明的环氧烷聚合催化剂,能够有效地制造聚环氧烷。所得聚环氧烷对于聚氨酯原料、聚酯原料、表面活性剂原料、润滑剂原料等而言是有用的。
此外,本发明的聚环氧烷对于聚氨酯原料、聚酯原料、表面活性剂原料、润滑剂原料等而言是有用的。
通过使用本发明的环氧烷聚合催化剂而得到的聚环氧烷尤其是与各种异氰酸酯化合物发生反应,可期待在绝热材料等中使用的硬质泡沫、汽车的坐垫/椅垫、床上用品等中使用的软质泡沫、粘接剂、涂料、密封材料、热固性弹性体、热塑性弹性体中展开应用。
- 还没有人留言评论。精彩留言会获得点赞!
- 氧化铝载体、其制备方法、由其制成的银催化剂及该银催化剂的应用的制作方法
- 环氧乙烷生产用银催化剂的载体、其制备方法、由其制成的银催化剂及其在环氧乙烷生产 ...的制作方法
- 一种氧化铝载体、其制备方法、由其制成的银催化剂及其应用的制作方法
- 环氧乙烷生产用银催化剂的载体、其制备方法、由其制成的银催化剂及其应用的制作方法
- 环氧乙烷生产用银催化剂的载体、其制备方法及其应用的制作方法
- 一种银催化剂的α-氧化铝载体的制备方法
- 生产环氧乙烷用银催化剂载体、制备方法及其应用的制作方法
- 一种乙烯氧化生产环氧乙烷用银催化剂的制作方法
- 一种银催化剂用多孔α-氧化铝载体的表面改性方法
- 氧化铝载体及其负载的银催化剂的制备方法及应用的制作方法