一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的精制方法与流程
本发明涉及高分子材料的精制领域,具体涉及一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的精制方法。
背景技术:
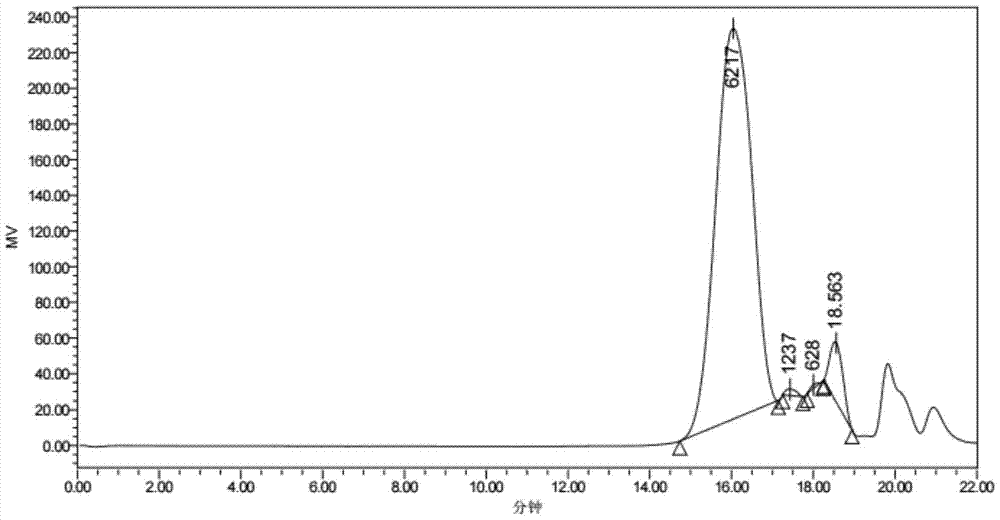
:聚乙二醇单甲醚-聚乳酸嵌段共聚物(mpeg-pdlla)是一种近年来被广泛研究的生物相容性好的生物可降解材料,一般是以聚乙二醇单甲醚(mpeg)和d,l-丙交酯(d,l-la)为单体,在辛酸亚锡的催化作用下进行配位开环聚合得到。该材料在体内可降解为聚乙二醇单甲醚和乳酸。其中mpeg具有优异的生物相容性,在体内能溶于组织液中,分子量4000以下的mpeg能被机体迅速排出体外而不产生任何毒副作用,其安全性已经得到了美国fda的认证。乳酸则可进一步降解为二氧化碳和水,聚合物的降解产物均不存在潜在的毒副作用。聚乙二醇单甲醚-聚乳酸嵌段共聚物(mpeg-pdlla)的临界胶束浓度(cmc)低,具有较好的物理稳定性、较高的载药量(李晓然,袁晓燕.聚乙二醇-聚乳酸共聚物药物载体[j].化学进展,2007,19(6).)。这种聚合物形成的核-壳结构的胶束,粒径在100nm以内(vladimirp.torchilin,structureanddesignofpolymericsurfactant-baseddrugdeliverysystems,journalofcontrolledrelease,2001,73:137-172.)。其亲水的peg外壳使聚合物胶束不易被网状内皮系统(res)所识别而清除,因而在血液中具有较长的驻留时间;而纳米级的粒径使之由于增强渗透滞留(epr)效应能在肿瘤部位积蓄,具有一定的被动靶向作用。2006年12月由韩国samyang公司研发的注射用胶束化紫杉醇在韩国上市,商品名为genexol-pm,随后在南亚一些国家上市,2009年向美国fda提出新药申请,目前在美国已经完成ⅱ期临床和ⅲ期临床第一阶段研究(kimsc,kimdw,shimyh,etal.invivoevaluationofpolymericmicellarpaclitaxelformulation:toxicityandefficacy[j].journalof controlledrelease,2001,72(1):191-202.)。然而目前生产、临床应用中发现存在一些不足,该制剂冻干粉及复溶溶液放置稳定性差,批次差别大,生产重现性不理想。研究表明聚合物材料的质量对生产应用及制剂稳定性有显著影响。中国专利申请cn103768013a公布了一种以精制两亲性嵌段共聚物(mpeg-pdlla)为载体的紫杉醇聚合物胶束,其中mpeg-pdlla的精制工艺是以阳离子交换树脂处理聚乙二醇单甲醚-聚乳酸嵌段共聚物,能够有效降低锡含量,使复溶后的胶束溶液稳定性明显增加。中国专利申请cn104892909a公开了一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的制备方法,反应完毕后的后处理方法采用1-4次二氯甲烷溶解-乙醚结晶过程,其中在第一次或第二次结晶过程中加入将待纯化产品用二氯甲烷溶解后加入纯水洗涤的操作,能够去除更多低分子杂质,使产品的分子量分布明显降低,产品吸湿性有了异常明显的改善,使流动性更好,更适合产品的保存和使用。中国专利cn102181048b公开了一种医用聚醚/聚酯嵌段共聚物的制备方法,反应结束后产物中残存的未反应单体通过加水反应去除,重金属催化剂通过高速离心的方法去除。利用这种共聚物载药有效地提高了难溶性药物的溶解度,改善了药物的安全性和有效性,但其缺点在于用水分散后的稳定性较差,在很短的时间内就出现了药物的泄露,使得在临床应用时因其物理稳定性不高而无法进一步推广和真正应用。为了解决胶束制剂的稳定性,中国专利cn101972480b公开了一种通过在聚合物胶束中添加氨基酸的方法来提高胶束复溶后稳定性的技术,但是添加的物质对于工业化生产要求较高,并且添加的稳定剂增加了制剂工艺的繁琐性,同时添加的氨基酸等对主药有着降解作用,不适合大规模生产。研究表明mpeg-pdlla的分子量均一性是影响胶束的大小和稳定性的重要因素。中国专利申请cn103980466a公布了一种法以阴离子聚合机理制备聚乙二醇-聚乳酸嵌段共聚物,分子量具有更好的可控性,分子量分布窄,反应温度低,副反应少。但是使用的催化剂萘钾不适用于药用领域。中国专利申请cn104761710a公布了一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的制备,单体在真空下熔融聚合,经过三次精制的聚合物分子量分布非常窄,制备的冻干制剂水分散后包封率大于90%的时间可以达到12小时以上,效果远远 优于普通的冻干制剂。但是精制过程并没有除去聚合物中分子量偏高的部分,会延长制剂制备时的溶解时间和冻干制剂的复溶时间,并且增大胶束溶液的粒径。技术实现要素:本发明的目的是提供一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的精制方法。通过逐步降温法对聚合物进行精制处理,使得聚合物的分子量分布缩窄,以该精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制备得到的聚合物胶束稳定性显著提高。本发明技术方案:一种聚乙二醇单甲醚-聚乳酸嵌段共聚物的精制方法,采用逐步降温法,包括步骤:(1)将聚乙二醇单甲醚-聚乳酸嵌段共聚物粗品与有机溶剂甲混合,在40℃-55℃使粗品充分溶解,然后降温至30℃-40℃静置,使混合物充分分层至上层澄清,收集上层清液;将上层清液温度降至10℃-20℃(优选为15℃-18℃)后静置析出沉淀,收集沉淀;(2)将步骤(1)中的沉淀用有机溶剂乙溶解,在冰乙醚或冰甲基叔丁基醚中沉淀,收集沉淀物,真空干燥,得到精制的聚乙二醇单甲醚-聚乳酸嵌段共聚物。为了达到更好的发明效果,优选:步骤(1)中,所述有机溶剂甲采用甲醇、乙醇、异丙醇中的一种,或者采用甲醇、乙醚、甲基叔丁基醚中的一种与乙腈、丙酮、二氯甲烷、三氯甲烷中的一种的两两组合。所述有机溶剂甲采用两两组合时可以采用任意比例,进一步优选为甲醇、乙醚、甲基叔丁基醚中的一种与乙腈、丙酮、二氯甲烷、三氯甲烷中的一种的体积比为1.5-10:1(最优选4-9:1)。所述每克聚乙二醇单甲醚-聚乳酸嵌段共聚物粗品中有机溶剂甲的用量为7ml-50ml。所述在40℃-55℃使粗品充分溶解步骤的温度高于降温至30℃-40℃静置步骤的温度。进一步优选所述在40℃-55℃使粗品充分溶解步骤的温度与降温至30℃-40℃静置步骤的温度之差至少为5℃(最优选至少为 10℃)。步骤(2)中,所述有机溶剂乙采用乙腈、二氯甲烷、氯仿或丙酮。所述冰乙醚或冰甲基叔丁基醚采用本领域常用的冰乙醚或冰甲基叔丁基醚,例如可选用2℃-8℃的乙醚或2℃-8℃的甲基叔丁基醚。所述真空干燥的温度不宜过高,一般为20℃-35℃较适宜。所述聚乙二醇单甲醚-聚乳酸嵌段共聚物粗品是以聚乙二醇单甲醚和d,l-丙交酯为单体,在辛酸亚锡的催化作用下进行配位开环聚合得到。可直接采用市售产品也可采用现有的制备方法制备(luckea,teβmarj,schnelle,etal.biodegradablepoly(d,l-lacticacid)-poly(ethyleneglycol)-monomethyletherdiblockcopolymers:structuresandsurfacepropertiesrelevanttotheiruseasbiomaterials[j].biomaterials,2000,21(23):2361-2370.)。其中聚乙二醇单甲醚的数均分子量为2000-10000,聚乙二醇单甲醚与d,l-丙交酯的质量比为1:0.5-1.2。聚乙二醇单甲醚数均分子量优选为2000,聚乙二醇单甲醚与d,l-丙交酯的质量比优选为1:0.8-1.0。制备方法包括:配方量的聚乙二醇单甲醚和d,l-丙交酯用甲苯共沸除水后加入催化剂辛酸亚锡,120℃-130℃反应22小时-26小时,反应完成后在乙醚或甲基叔丁基醚中搅拌沉降,真空干燥得聚合物粗品。所述聚乙二醇单甲醚-聚乳酸嵌段共聚物粗品的分子量分布一般在1.2以上,通过本发明逐步降温法精制能够得到分子量分布为1.15以下的聚乙二醇单甲醚-聚乳酸嵌段共聚物。本发明的有益效果:本发明逐步降温法精制能够得到分子量分布为1.15以下的聚乙二醇单甲醚-聚乳酸嵌段共聚物,聚合物的分子量分布缩窄。采用本发明精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制备得到的胶束粒径小且均一,冻干粉稳定性优异,复溶效果好。以该精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制备得到的紫杉醇聚合物胶束稳定性显著提高。本发明方法工艺流程简单,用时短且适宜放大生产。附图说明图1为实施例1中mpeg-pdlla粗品的gpc图谱;图2为实施例2中mpeg-pdlla粗品的gpc图谱;图3为实施例3中mpeg-pdlla粗品的gpc图谱;图4为实施例4中精制后mpeg-pdlla的1h-nmr图谱;图5为实施例4中精制后mpeg-pdlla的gpc图谱;图6为实施例5中精制后mpeg-pdlla的gpc图谱;图7为实施例6中精制后mpeg-pdlla的gpc图谱;图8为实施例7中精制后mpeg-pdlla的gpc图谱;图9为实施例8中精制后mpeg-pdlla的gpc图谱;图10为实施例9中精制后mpeg-pdlla的gpc图谱;图11为实施例10中精制后mpeg-pdlla的gpc图谱。具体实施方式下面将通过具体实施例对本发明做进一步说明,但需要指出的是,以下实施例不能构成对本发明的任何限制。聚乙二醇单甲醚-聚乳酸嵌段共聚物的粗品和精制产品采用核磁氢谱和凝胶渗透色谱(gpc)进行分子量及分子量分布分析。实施例1称取mpeg-200050.1g、d,l-la45.5g和甲苯500ml加入反应釜中,搅拌加热升温至110℃,共沸去水。然后停止加热,将混合物的温度降至低于90℃,加入0.25g的催化剂辛酸亚锡。在搅拌下,混合物保持130℃反应23小时。反应完成后降温至50℃,在4℃冰乙醚中搅拌沉降,收集沉淀物,真空干燥得mpeg-pdlla粗品。图1的gpc检测结果如下:mn:5474,分子量分布:1.227。其中,表1为图1中宽分布未知样相对峰表。表1实施例2称取mpeg-200049.6g、d,l-la46.1g和甲苯500ml加入反应釜中, 搅拌加热升温至120℃,共沸去水。然后停止加热,将混合物的温度降至低于90℃,加入0.25g的催化剂辛酸亚锡。在搅拌下,混合物保持120℃反应22小时。反应完成后降温至60℃,在4℃冰乙醚中搅拌沉降,收集沉淀物,真空干燥得mpeg-pdlla粗品。图2的gpc检测结果如下:mn:5588,分子量分布:1.206。其中,表2为图2中宽分布未知样相对峰表。表2实施例3称取mpeg-200050.4g、d,l-la42.1g和甲苯500ml加入反应釜中,搅拌加热升温至130℃,共沸去水。然后停止加热,将混合物的温度降至低于90℃,加入0.25g的催化剂辛酸亚锡。在搅拌下,混合物保持130℃反应24小时。反应完成后降温至60℃,在4℃冰甲基叔丁基醚中沉降,收集沉淀物,真空干燥得mpeg-pdlla粗品。图3的gpc检测结果如下:mn:5091,分子量分布:1.313。其中,表3为图3中宽分布未知样相对峰表。表3实施例4取实施例1的mpeg-pdlla粗品10.02g,加入200ml甲醇,在40℃下搅拌40min,搅拌速度为60r/min,使粗品充分溶解;然后缓慢降温至30℃静置,使混合物充分分层至上层澄清,在温度设置为30℃的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在15℃下静置60min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后 再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率35%。得到的聚合物用氢核磁共振和凝胶渗透色谱进行表征,结果如图4、表4和图5。图4为聚乙二醇单甲醚-聚乳酸嵌段共聚物的核磁氢谱表征。pdlla段分子量为1883,符合单体投料比。图5的gpc检测结果如下:mn:5836,分子量分布:1.105。其中,表4为图5中宽分布未知样相对峰表。表4实施例5取实施例2的mpeg-pdlla粗品10.14g,加入80ml乙醇至反应釜中,在50℃下搅拌40min,搅拌速度为60r/min,使粗品充分溶解;然后缓慢降温至40℃静置,使混合物充分分层至上层澄清,在温度设置为40℃的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在18℃下静置60min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率36%。得到的聚合物用凝胶渗透色谱进行表征,结果如图6和表5。图6的gpc检测结果如下:mn:5757,分子量分布:1.103。其中,表5为图6中宽分布未知样相对峰表。表5实施例6取实施例2的mpeg-pdlla粗品10.01g,加入100ml乙醇至反应釜中,在52℃下搅拌40min,搅拌速度为60r/min,使粗品充分溶解;然后缓慢降温至40℃静置,使混合物充分分层至上层澄清,在温度设置为40℃ 的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在18℃下静置60min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率33%。得到的聚合物用凝胶渗透色谱进行表征,结果如图7和表6。图7的gpc检测结果如下:mn:5877,分子量分布:1.119。其中,表6为图7中宽分布未知样相对峰表。表6实施例7取实施例2的mpeg-pdlla粗品10.05g,加入150ml乙醇至反应釜中,在55℃下搅拌40min,搅拌速度为60r/min,使粗品充分溶解;然后缓慢降温至40℃静置,使混合物充分分层至上层澄清,在温度设置为40℃的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在18℃下静置60min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率31%。得到的聚合物用凝胶渗透色谱进行表征,结果如图8和表7。图8的gpc检测结果如下:mn:5968,分子量分布:1.097。其中,表7为图8中宽分布未知样相对峰表。表7实施例8取实施例2的mpeg-pdlla粗品10.11g,加入200ml乙醇至反应釜中,在50℃下搅拌40min,搅拌速度为60r/min,使粗品充分溶解;然后缓慢降温至40℃静置,使混合物充分分层至上层澄清,在温度设置为40℃ 的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在18℃下静置60min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率27%。得到的聚合物用凝胶渗透色谱进行表征,结果如图9和表8。图9的gpc检测结果如下:mn:6040,分子量分布:1.097。其中,表8为图9中宽分布未知样相对峰表。表8实施例9取实施例2的mpeg-pdlla粗品10.07g,加入300ml乙醚-三氯甲烷混合溶剂(乙醚与三氯甲烷的体积比为4:1)至反应釜中,在50℃下搅拌30min,搅拌速度为30r/min,使粗品充分溶解;然后缓慢降温至30℃静置,使混合物充分分层至上层澄清,在温度设置为30℃的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在15℃下静置30min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率36%。得到的聚合物用凝胶渗透色谱进行表征,结果如图10和表9。图10的gpc检测结果如下:mn:6012,分子量分布:1.095。其中,表9为图10中宽分布未知样相对峰表。表9实施例10取实施例3的mpeg-pdlla粗品10.23g,加入500ml甲醇-二氯甲烷混合溶剂(甲醇与二氯甲烷的体积比为9:1)至反应釜中,在45℃下搅拌 30min,搅拌速度为30r/min,使粗品充分溶解;然后缓慢降温至35℃静置,使混合物充分分层至上层澄清,在温度设置为35℃的离心机中离心,转速1700r/min,离心5分钟,收集上层清液;将上层清液在16℃下静置30min析出沉淀后离心,收集下层沉淀;将沉淀用适量丙酮溶解后再在4℃冰甲基叔丁基醚中沉淀,离心收集沉淀物,30℃真空干燥,得到精制后的mpeg-pdlla。收率33%。得到的聚合物用凝胶渗透色谱进行表征,结果如图11和表10。图11的gpc检测结果如下:mn:5481,分子量分布:1.092。其中,表10为图11中宽分布未知样相对峰表。表10实施例11紫杉醇聚合物胶束冻干制剂的制备和稳定性考察1、利用本发明制备的嵌段聚合物为载体制备药物纳米聚合物胶束(1)取紫杉醇300mg,实施例制备的mpeg-pdlla粗品及精制后mpeg-pdlla各1500mg,有机溶剂丙酮30ml,备用。(2)向mpeg-pdlla和紫杉醇的混合物中加入30ml丙酮摇晃溶解,然后在50℃、100r/min的转速下旋蒸1h,蒸去丙酮得到紫杉醇和聚合物混合凝胶膜,快速加入50ml50℃的去离子水,涡旋至完全水化得胶束溶液,然后0.22μm滤膜过滤,分装成5ml每瓶,冻干。2、以精制前后聚合物制备的胶束冻干粉进行稳定性考察精制前后聚合物制备的胶束冻干制剂在室温25℃下干燥器内放置15天后各取1瓶,以生理盐水5ml复溶,考察了复溶时间、粒径及分布。将复溶后的制剂溶液在室温25℃下放置24小时,监测了粒径及分布和包封率的变化。结果见表11和表12。表11紫杉醇胶束制剂的复溶时间、溶剂的粒径及分布结果表12紫杉醇胶束溶液包封率随时间的变化结果表明,以本发明精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制得的紫杉醇聚合物胶束冻干粉复溶时间短,复溶后的粒径稳定性比精制前聚合物(即粗品)制得的紫杉醇聚合物胶束更好。以本发明精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制得的紫杉醇聚合物胶束冻干粉复溶后24小时胶束溶液仍保持淡蓝色澄清透明,紫杉醇的包封率基本保持不变。实施例5-8和实施例10中精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制得的紫杉醇聚合物胶束冻干粉复溶时间短,复溶后的粒径稳定性更好,复溶后24小时胶束溶液仍保持淡蓝色澄清透明,紫杉醇的包封率基本保持不变。中国专利cn103768013a涉及的纯化方法能有效降低聚合物中的锡含量,以其精制前后的聚合物制备得到的紫杉醇聚合物胶束溶液粒径随时间的变化如下表13:表130hr6hr10hr15hr24hr精制前粒径nm30.2±0.429.2±0.428.8±0.540.8±1.4白色悬浮物精制后粒径nm29.5±0.528.7±0.428.0±0.329.8±0.538.5±0.1数据显示,以本发明精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制 得的紫杉醇聚合物胶束冻干粉复溶时间短,复溶24小时后的粒径稳定性更好。本发明精制方法中各参数的变化(如数据、有机溶剂种类的变化)均能够得到分子量分布为1.15以下的聚乙二醇单甲醚-聚乳酸嵌段共聚物精制品,以该精制后的聚乙二醇单甲醚-聚乳酸嵌段共聚物制备得到的紫杉醇聚合物胶束粒径小且均一,冻干粉稳定性优异,复溶效果好。因此本发明制备方法中任意参数的组合均可实现本发明的目的。在此不再赘述。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1