游离脂肪酸在水存在下向乙酯的转化的制作方法
本申请要求于2015年6月24日提交的美国临时申请号62/184,064的优先权,该临时申请通过引用为了所有目的完整地结合于此。
技术领域:
本发明涉及一种在水存在下将海藻油转化为脂肪酸乙酯(FAEE)的方法。
背景技术:
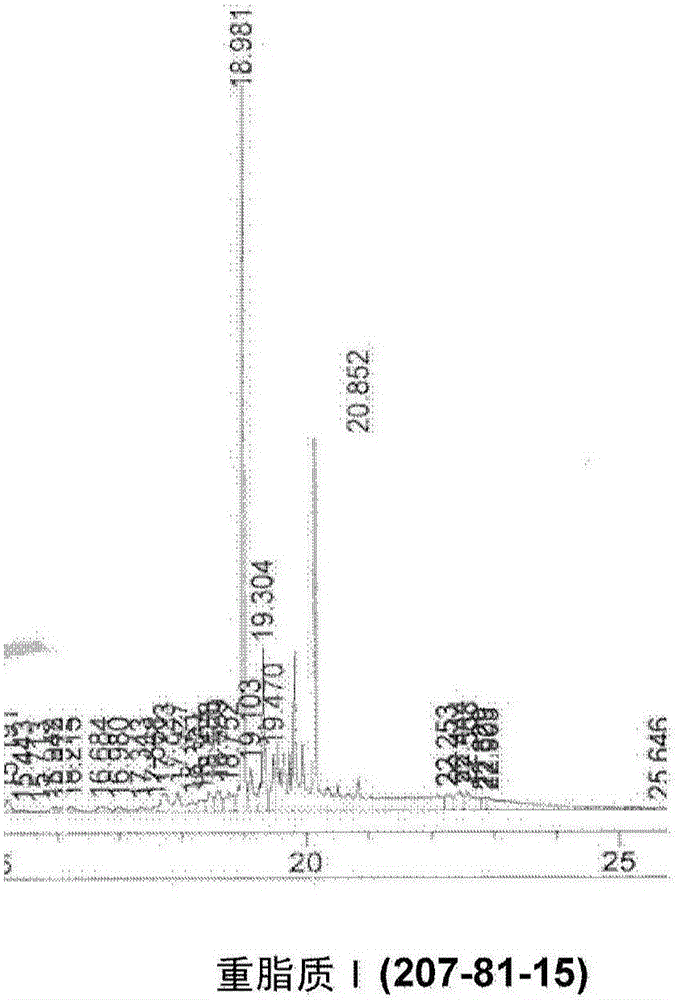
:在与脂肪酸、脂肪酸盐或脂肪酸酯的典型酯化和酯交换反应中,反应混合物中的任何水导致化学效率的大量损失。那是因为水将(相对于醇)优先与脂肪酸反应而产生游离脂肪酸,而非所需酯。多年来已经开发多种技术以从反应混合物中去除水,以便实现酯的高转化率。标准技术是从进料中消除水,使用醇盐碱,和/或从酯化反应中汽提水。技术实现要素:本文提供了一种将海藻油在水存在下转化为脂肪酸乙酯(FAEE)的方法。该方法包括使海藻油与碱金属碱(alkalibase)和醇反应以生产皂化的粗油;将浓硫酸添加至皂化的粗油中以生产游离脂肪酸(FFA);和将乙醇添加至FFA中以转化为FAEE。在一些实施方案中,碱金属碱是NaOH或KOH。在一些情形中,海藻油包含至少约1-10%的水(按重量计)。在一些情形中,将大约0.4-50%浓硫酸(按重量计)添加至皂化的粗油中。在一些情形中,将大约3-20%乙醇(按重量计)添加至FFA中。海藻油可以在皂化反应器中反应。FFA可以在皂化反应器中转化为FAEE。根据下列详述和附图,本发明的其他目的、特征和优势对于本领域技术人员来说将会是显而易见的。附图说明图1显示了本文提供的方法的一个示例性实施方案。图2A和2B显示对于级分I(图2A)和级分II(图2B)的重脂质GC峰。图3显示相对于逐渐降低的pH,酯转化的增加。图4显示相对于逐渐降低的pH,水浓度的增加。图5提供对于连续转化运行的步骤1的工艺流程图。图6提供对于连续转化运行的步骤2的工艺流程图。图7显示最终转化产物(粗FAEE)中的脂质组合物。图8显示良好皂化和不良皂化样品色谱图。图9提供在皂化期间发生的反应的图解。图10显示皂化样品和完全反应的相同样品的色谱图。图11显示在酯化之前和之后的样品的色谱图。图12显示来自步骤1和2的Karr柱的废物流的色谱图。图13显示在热箱加入(hotboxaddition)之前和之后Karr塔顶馏出物(overhead)和尾分(tail)两者的挥发物的百分比。图14显示相对于进料的pH,Karr塔顶馏出物和尾分两者的挥发物的百分比。图15显示相对于百分比转化,混合酯化样品中水的量。图16显示在庚烷汽提的最后两天期间换热器出口的温度。图17显示正常的酯化样品(左)和浅色酯化样品(右)。图18显示Karr柱进料,塔顶馏出物和尾分的色谱图。图19显示水洗进料,塔顶馏出物和尾分的色谱图。具体实施方式本文公开的方法可以用于实现酯交换反应中游离脂肪酸(FAA)向脂肪酸乙酯(FAEE)的高转化率(例如,至少96%转化),尽管存在大约1%-20%的水(按重量计),例如,大约1%-20%,1%-5%,5%-20%,1%-15%,5%-15%,1%-10%,10-20%,5%-10%,10%-15%,或15%-20%的水(按重量计)。该方法部分基于以下出人意料的发现:当海藻油被酸化至2.0以下的pH时,当乙醇:水比率为大约5时,可以实现至少96%的高转化率。在一些实施方案中,使用浓硫酸将海藻油酸化。在一些情形中,酸化的海藻油具有2.0以下的pH,例如pH2,pH1.9,pH1.8,pH1.7,pH1.6,pH1.5,pH1.4,pH1.3,pH1.2,pH1.1,pH1.0,或更小。在不限于具体理论的情况下,酸浓度降低相对于乙醇的水逸度(waterfugacity),导致高转化率。无机酸如硫酸需要水分子来充分水合。因此,酸的水合可能会降低水分子与脂肪酸部分反应的可获得性并且阻碍酯交换反应。在本文提供的方法中,存在海藻油至脂肪酸乙酯(FAEE)的两步转化。海藻油可以含有大量的水,如大约1至大约20%的水(按重量计)或更多,例如,大约1%,大约2%,大约3%,大约4%,大约5%,大约6%,大约7%,大约8%,大约9%,大约10%,大约11%,大约12%,大约13%,大约14%,大约15%,大约16%,大约17%,大约18%,大约19%,大约20%,或更多水(按重量计)。在一些实施方案中,海藻油具有大约1%-20%的水(按重量计)或更多,例如,大约1%-20%,1%-5%,5%-20%,1%-15%,5%-15%,1%-10%,10-20%,5%-10%,10%-15%,或15%-20%或更多水(按重量计)。在方法的第一步中,海藻油与乙醇和碱金属碱如NaOH或KOH反应。在一些情形中,将大约3-20%的乙醇(按重量计),例如,大约3%,大约4%,大约5%,大约6%,大约7%,大约8%,大约9%,大约10%,大约11%,大约12%,大约13%,大约14%,大约15%,大约16%,大约17%,大约18%,大约19%,或大约20%乙醇(按重量计)加入至海藻油。在一些实施方案中,加入大约5-35%的NaOH(按重量计),例如,大约5%,大约5%,大约10%,大约15%,大约20%,大约25%,大约30%,或大约35%的NaOH(按重量计)。在其他实施方案中,加入大约5-35%的KOH(按重量计),例如,大约5%,大约5%,大约10%,大约15%,大约20%,大约25%,大约30%,或大约35%的KOH(按重量计)。该第一步(其可以在皂化反应器中进行)将所有脂肪酸部分转化为它们的碱金属皂(alkalisoap)或乙酯形式。在第二步中,用浓硫酸将皂化的粗油酸化。在一些情形中,加入大约0.4-50%的浓硫酸(按重量计),例如,大约0.4%,大约0.6%,大约0.8%,大约1%,大约5%,大约10%,大约15%,大约20%,大约25%,大约30%,大约35%,大约40%,大约45%,或大约50%的浓硫酸(按重量计)。硫酸将皂转化为游离脂肪酸(FFA)形式。FFA然后可以通过与乙醇反应催化而转化为乙酯(FAEE)。在过量水存在下,FFA向FAEE的转化不是高效的,因为水对油比乙醇更具有反应性。然而,在过量酸(pH<2.0)的存在下,FFA以极高的转化率,如大于95%,例如,96%,97%,98%,99%,或更高,转化为FAEE。本文公开的方法避免了以下需要:从酯化反应器中汽提乙醇/水混合物,从该物流中去除水,和/或将干燥乙醇返回反应器。该方法允许在皂化反应器中使用海藻油。另外,该方法利用碱的氢氧化物形式而不是乙醇盐形式。该高转化率的新方法比将进料干燥或将反应混合物干燥明显更便宜和更容易。实施例实施例1:海藻油乙酯工艺研究介绍和背景:数年前,Aurora,Inc.开发了一种化学工艺从己烷萃取的海藻油中生产脂肪酸甲酯(FAME’s)。该工艺首先在Aurora实验室中逐步定义,然后在中试工厂(pilotplant)中论证。该工艺成功地从该海藻油中生产FAME’s。Aurora随后的研究确定了藻类的己烷萃取是不充分的。己烷只是过于非极性的萃取溶剂,在藻类中留下大量的极性脂质,含有大量EPA的物质,高度评价的FFA。尽管仍然使用己烷萃取,但是Aurora认识到将甲醇加入己烷改善了这些所需极性脂质的回收,但不如完全更换为乙醇。因此,乙醇成为选择的萃取溶剂。更换为乙醇导致之前为制备FAME’s开发的工艺中的一些变化。其一,它为工艺简化提供了一个大机会。FAME’s从来都不是Aurora’s所要的最终产品。它们将被酯交换为它们的乙酯(FAEE’s-脂肪酸乙酯),从其中分离EPA乙酯用于商业销售。并且MATRIC已经定义了一种用于该转化的有效反应蒸馏工艺。但是现在,在乙醇已经存在于萃取的油中的情况下,本身有机会直接制备乙酯。该工作是本报告的焦点。部分I.直接酸催化的酯化/酯交换用于确定脂质中皂化当量的ASTM分析步骤之一利用在酸性条件下该脂质的总酯化。一种技术利用甲醇中的乙酰氯;另一种利用甲醇中的浓硫酸。利用这些步骤,我们认为值得尝试在一个酯化/酯交换步骤中由含乙醇的海藻油直接制备乙酯。仅仅使用乙醇、硫酸和足够时间和温度,我们能够完全将海藻油中的所有脂质酯交换为它们的乙酯吗?如果使用硫酸和甲醇能够实现脂质定量转化,使用硫酸和乙醇是否也能够?如果是这样,我们能够大幅简化该工艺。实验室运行首先表明这也许是可能的,因为我们使用的一些较新的GC方法(以实现EPA乙酯和EPA本身之间的基线分离)显示在最终GC扫描中“没有一个”重脂质出现并且显示我们通常遇到的所有FFA’s的高乙酯浓度。不幸地,这是一个假象,因为许多未反应的重脂质并未送到柱子上,而是保留在GC的注入口。这通过使用柱上(on-column)注入很快证实,其中我们可以看到重脂质,但是也许并非它们所有。然而,即使柱上分析也表明更多的时间、和/或温度和/或更多的硫酸也许能够实现剩余重脂质向所需乙酯的越来越深入的转化。因此,我们进一步研究。尽管我们能够通过该方法将很多,也许甚至大部分藻类脂质直接转化为乙酯,但是很明显仍然存在极性的含EPA的藻类脂质,其抵制酸催化的转化并且保持未转化。另外,实现更深入的脂质转化所需的极端反应条件可能将一些存在的乙醇转化为二乙醚并且也可能将一些EPA异构化为其反式异构体。在中试工厂尝试该一步工艺后,不得不放弃直接酸催化酯交换为乙酯,因为海藻油脂质的转化不充分。乙醇-硫酸混合物中的水形成。在海藻油直接转化为乙酯中使用硫酸的另外的挑战之一是决定如何将硫酸加入反应混合物。不想将浓硫酸直接加入乙醇-海藻油,我们选择将其与无水乙醇预混合,然后将乙醇硫酸加入海藻油反应混合物。这防止将海藻油直接暴露于浓酸。但是,在研究在乙醇中在不同温度下制备各种浓度的硫酸时,我们发现这实际上是一个非常具有挑战性的操作。无水乙醇和浓硫酸几乎立即开始彼此反应,产生硫酸单乙酯(硫酸氢乙酯),水和热。这是一个平衡反应,平衡的位置和达到平衡的速率取决于反应物/产物的浓度和温度。对于更浓的溶液,需要冷却来去除在将硫酸加入乙醇过程中混合/反应产生的热。尽管我们最终放弃了该一步工艺,但是来自该研究的发现有助于我们在制备用于酯化的乙醇-硫酸混合物时将水形成最少化。实验:我们研究了硫酸氢乙酯形成的速率,从无水乙醇中24重量%的硫酸开始,接着增加化学计量副产物水的浓度。在每个温度下的实验期间,将硫酸浓度逐步升至33%,42%和最终50%的酸。我们在0℃,“室温”和40℃进行这些反应。选择这些硫酸浓度以帮助中试工厂决定用于稀释反应器的目标浓度和确定是否将需要冷却。通过将25.0g无水乙醇带至目标温度在125ml锥形瓶(Erlenmeyerflash)中进行反应。利用磁力搅拌并在大约4-5分钟的时间段内加入8.0g的浓硫酸,该速率不会使得溶液温度上升太快。周期性地取出样品用于立即Karl-Fisher分析,并且继续反应直至观察到稳态浓度的水。然后,加入下一个增量的硫酸,重复该过程。记得浓硫酸名义上是94-96%,因此,一些水直接来自所加入的酸。因此,我们寻找水浓度相对于该初始基线水水平的增加,以确定何时硫酸氢乙酯形成变得明显。结果在下表中给出。在大约1℃收集的这些数据显示,硫酸氢乙酯和水的形成在30分钟时间范围内相当慢。仅在50%酸浓度下似乎水从15至30分钟有显著增加–并且这如果是真的话,是轻微的。可以在这些条件下进行稀释反应,并且不产生明显的硫酸氢乙酯和水,如果在该30分钟时间范围内消耗混合物的话。名义室温下的这些数据显示,在酸加入后15-30分钟期间,即使在24.6%酸浓度下,水浓度也缓慢增加。尽管该增加在较低浓度下是轻微的,但是每个30分钟的值大于其15分钟的对应物。另外,随着酸强度增加,这些增加变得更大。保持50%酸样品另外15分钟以阐明这一点。通过检验40℃的数据并将其与先前数据比较,我们认识到反应已经很好地进行,即使在15分钟选取第一个样品之前和24%酸下。该水浓度大于在该相同反应时间在前两个0℃和“室温”运行中水浓度的两倍大。该水浓度在15-30分钟点期间仍然增加,但是仅仅缓慢增加,因为反应趋近平衡。不推荐在这些条件下操作稀释反应器。幸运地,在这些运行中没有一个检测到二乙醚(DEE)。并且这真的没有预料到。文献告诉我们通过硫酸酯途径的DEE形成只有到了高得多的温度和更无水的条件下才变得显著。在那些条件下,甚至可以制备硫酸二乙酯。硫酸二乙酯是一个问题,因为其具有毒性并且其形成应当避免。当足够的水存在时,它不会以任何明显的程度形成。直接酸催化的酯交换:实验和结果:在“回流”下的直接5%酸催化的酯交换。我们首先估计该一步转化通过将海藻油和乙醇的溶液在5重量%硫酸存在下回流(~90℃)的可能性。转化后接着对以周期间隔选取的样品进行GC分析。从充分搅拌的反应混合物中去除样品(~0.5g),与5ml己烷混合并离心10-20分钟。这允许酸分离至极性重相(水,甘油,乙醇等),并且顶部己烷层的样品可以直接衍生化和通过GC分析。对于该初始工作,我们的确利用“柱上”分析。反应进行后接着观察C16FFA’s和EPA的酯化和在色谱图的19.5-22.5分钟区域中总和的“重脂质峰”的消失。我们还观察了在22.4分钟的尖峰(据信是C16FFA’s的甘油三酯)的消失。在转化后使用色谱图的该“重脂质”区域来不是一个绝对措施,因为存在也在色谱图的该区域内出现的一些不可皂化的组分。还可能的是,该积分窗口(integrationwindow)可能不是足够宽的,因为我们已经观察到保留时间偏移和有时甚至已知的脂质峰,如22.4分钟峰,可以落在该窗口之外。然而,这是用于估计脂质转化率的有用工具。然而,它证明对于确定总脂质转化是不够的。发现存在的FFA’s的酯化比重脂质的转化要快得多。该第一个实验的结果显示如下。在“回流(90℃)”下的直接酸处理(5%)结果。样品时间(hr.)C16乙酯选择性EPA乙酯选择性重脂质*10.2558.654.621.8-4.3920.7587.180.415.6-2.3731.7591.474.37.41-1.4242.7593.892.64.79-1.0753.7592.990.64.90-0.7864.7593.991.83.45-0.98*第一个值是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图是207-50-1至207-50-6。根据该GC数据,很清楚这些条件是可以将重脂质转化为它们的乙酯,但是该转化要花费大量时间。还清楚的是,“酯交换”是比直接酯化FFA’s更慢的反应。另一个问题是,难以知道究竟有剩余多少未转化的脂质。尽管GC扫描到达其中在22.4分钟的“重脂质”峰和TG峰似乎停止减小的点,但是仍然可能存在其他含EPA的脂质,它们仅仅没显示在GC色谱图中或在该VVA+窗之外。然而,这些初始的实验室试验是足够以有希望的,我们进一步通过研究酸浓度、温度、时间和乙醇浓度的其他组合是否可以成功实现总脂质转化来继续该一步工艺。这些研究总结在以下描述中。“室温”下直接酸催化的酯交换(5%酸)该下一个实验是意欲看看乙醇萃取的和含乙醇的海藻油是否也许可以在贮存槽中反应(或“预反应”)并且使得更易于在更强迫的条件下的后面的总转化。之前使用FAME’s系统的研究已经教导我们,在过夜室温反应期间,直接FFA酯化容易进行至完全。因此,我们确信我们将会至少酯化海藻油中存在的所有FFA’s。我们也能够转化重脂质吗?为此,使得海藻油(5g)和无水乙醇(5g)的混合物与由0.7g硫酸和2.9g的无水乙醇制备的硫酸溶液在室温搅拌(磁力混合)。这得到5重量%酸溶液,乙醇与海藻油的比率为大约2。这考虑了海藻油本身含有大约20%残余的乙醇。由于这意欲评估在贮存槽条件下的转化,所以使得该反应进行一周的时间。周期性地将0.5g样品去除,与5ml己烷混合,并离心以从酸和极性组分中分离非极性组分。己烷级分的GC柱上分析(衍生化方法)给出下列结果。在“室温(RT)”的直接酸处理(5%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图是207-53-1至207-53-3。这些结果是相当鼓舞人心的,并且支持以下假设:在槽中的贮存条件下能够实现显著转化,所述槽能够耐受酸性环境并且能够提供一些搅拌。认识到贮存槽不一定必须是在“室温”,酸浓度也不一定必须固定在5%,我们进一步在数个另外的温度/酸组成下研究直接酸转化。这些结果总结在下表中。在40℃的直接酸处理(1.33%酸)结果。样品时间(hr.)C16乙酯选择性EPA乙酯选择性重脂质*1111.86.825.6-3.62312.45.426.6-3.83511.56.526.9-3.84*+24小时10.25.026.3-3.95**+23天96.190.27.9-0.35*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是关于样品在它们随时间而增加时的207-60-1,207-60-10,207-61-3,207-61-6和207-61-7。4*-在RT;5**-在RT但是对于最后23天酸重构为5.2%。显然,1.33%酸对于该反应无所作为。我们不仅没有转化任何重脂质,我们甚至不能酯化原油中存在的FFA’s。这与之前的发现(一向显示由于其中的非脂质组分,海藻油能够“中和”阈限量的强酸)一致。分析该海藻油的超过2%的氮含量,即碱性蛋白、氨基酸和叶绿素的混合物,并且这些化合物中的“碱性”氮能够与强酸反应,转变为“N-H+”物质,其现在是太弱的酸而不能催化我们所需的化学作用。只有当我们超过该“阈限”,我们才能期望转化开始。1.33重量%的硫酸明显不足。该具体表中的最后一个条目是通过将额外的硫酸加入反应混合物使其达到5.2%酸和然后使其在室温搅拌另外23天而获得的。此后,我们观察到高脂质转化和高酯/FFA比率,即酯化平衡。在40℃的直接酸处理(2.66%酸)结果。样品时间(hr.)C16乙酯选择性EPA乙酯选择性重脂质*1122.910.826.2-4.12326.08.226.4-4.03550.827.026.7-4.04*+24小时74.345.621.6-3.75**+23天96.397.05.9-0.25*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是关于样品在它们随时间而增加时的207-60-6,207-60-9,207-61-2,207-61-5和207-61-8。4*-在RT;5**-在RT在其重构为5%酸之后。从该运行可以得出几个结论。首先,2.66%酸是刚好足够的酸以超过以上讨论的“阈限”并且开始缓慢催化所含FFA’s的直接酯化,但是这不是足以开始重脂质的任何显著转化的酸。类似于之前在1.33%酸下的运行,将反应混合物重构至5.2%酸并且使其静置和搅拌另外23天使得它获得与之前运行相同的转化状态。在40℃的直接酸处理(5.2%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-60-5,207-60-8,207-61-1,和207-61-4。4*-在RT。在40℃使用5.2%酸在24小时点的结果类似于在室温的那些。在50℃的直接酸处理(5.2%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-69-3至207-69-11。FFA酯化速率在50℃增加,但是重脂质转化仍然非常缓慢。在70℃的直接酸处理(3.7%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-70-2至207-70-14(仅偶数)和207-71-2。再次,FFA酯化进行但是重脂质仅非常缓慢地转化。在70℃的直接酸处理(5.1%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-70-1至207-70-13(仅奇数)和207-71-1。在70℃下5%酸在4小时内实现了95%乙酯选择性,也发生了重脂质的显著转化。然而,重脂质的减少在开始2小时内最初较快29%->20%,然后减慢。这可能是由于剩余的不同类型的残余重脂质之间的反应速率不同。硬脂酸酯可能是最后被转化的那个。在70℃的直接酸处理(5.1%酸)结果(重复:新海藻油样品)*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-77-5,207-77-7,207-77-10,207-77-14,207-78-2,207-78-5,207-78-8和207-78-11。在70℃的直接酸处理(6.3%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-77-6,207-77-9,207-77-12,207-77-15,207-78-3,207-78-6,207-78-9和207-78-12。参见该表中的数据,表明存在样品标记的混乱或反应的不均匀取样。在70℃的直接酸处理(7.1%酸)结果。*第一个数字是19.5和22.5分钟之间的面积百分比的总和(色谱图上VVA+),第二个数字是22.4分钟尖峰(据信是C16FFA’s的TG)的面积%。这里提到的GC色谱图分别是207-77-8,207-77-11,207-77-13,207-78-1,207-78-4,207-78-7,207-78-10,207-78-13。这些最后三个实验的结果与之前运行的那些不一致。它们表现得似乎存在的酸比我们已经物理加入的要少。这使得我们疑惑。进一步研究,我们发现用于这最后三次运行的海藻油与用于之前运行的那个“不同”。当从5加仑桶中去除海藻油时,整个桶始终被熔化并且混合以获得样品,其然后与另外的乙醇充分混合并且立即分成等分试样用于各个较早的运行。因此,我们相信,所有海藻油样品合理地是相同的。然而,对于这最后三个运行,海藻油在于等重量的乙醇混合后,静置在实验台顶部数小时,之后分成三分之一用于70℃的三个运行。该50/50海藻油-乙醇混合物的溶液/悬浮液(再混合前)含有比之前批次更多的沉淀物。怀疑该“沉淀物”含有比我们已经证明在海藻油中存在的更多的“吃酸(acid-eating)”组分。这可以解释为何这些运行似乎是“低酸”运行。因此,这些最后三个运行不能与较早的运行比较。然而,它们可以彼此比较,因为所用的海藻油在这些最后三个70℃运行之间具有代表性。似乎较高的酸运行(7.1%)比其在5.1和6.3%酸下的两个相伴运行更差。尽管难以确定(因为溶液如此非常深),但是我们从其他实验获知在这些反应中存在两个液相。硫酸主要存在于重质底相。我们还认识到存在的硫酸越多,其从顶部油相中拖入其自身的乙醇越多。因此,在70℃下利用7.1%酸看到的差的结果可能是由于油中乙醇的减少。在该乙醇-油相中可溶的酸的浓度也将下降。这两者的发生都将降低反应速率,情形似乎正是这样。尝试更好地理解乙醇萃取的海藻油中的“污泥”:已经观察到即使在将其进一步用等量的额外乙醇稀释后,在我们的乙醇海藻油中仍然存在“不溶物”,我们计划实验来确定这些“不溶物”是否含有有价值的脂质。在室温下完成最初稀释,因此在加入额外的乙醇后,获得的溶液将仅为大约37.5%非挥发物(von-volatile)。我们使用该稀释,因为中试工厂正进行相同稀释,使得进料更容易处理和泵送。将75.0克乙醇萃取的海藻油与75.0g的无水乙醇混合并且在室温充分混合。然后离心20分钟并且分成较轻上层(99.95g)和较稠污泥样底层(39.20g)。“消失”的物质作为离心管中的残余物丢失。对于两个级分确定总挥发物分析。轻级分含有34%非挥发物;较重级分含有43%。为了确保每个级分中的非挥发物将见到相同相对量的乙醇,将11.1g额外的乙醇加入至较重级分。现在每个级分含有34%非挥发物。将在20.01g的无水乙醇中的4.65克硫酸加入至较轻级分,并且将9.74g的无水乙醇中的2.24g硫酸加入至较重级分。因此,每个级分具有相同的乙醇:非挥发物比率和相同的酸:非挥发物比率(14%)。在并行反应中,反应性的唯一区别应当是非挥发物级分的差异的结果。由首先在70℃度过一小时组成的实验意欲将所有放回到“溶液”中,因为其将会在海藻油的热乙醇萃取之后。该运行的其余部分是在100℃。周期性地取出样品,与5ml己烷混合并离心,产生直接适于GC分析(使用衍生化和柱上注入)的顶层。结果在下表中给出。在100℃的直接酸处理轻/重海藻油级分结果。*轻海藻油级分的C16’s酯选择性(C16’s面积百分比的总和)**重海藻油级分的C16”s酯选择性(C16’s面积百分比的总和)GC扫描从207-81-1至207-81-16;对于I为奇数;对于II为偶数。反应第一小时为70℃,反应其余部分为100℃。从这些数据(图2A和2B)可以得出几个有趣的观察/结论。重脂质GC峰(VVA+峰=19.5和22.5min之间面积百分比的总和)对于重质、污泥样的海藻油级分II开始高大约4%并且结束时同样高4%。GC扫描检查显示,这主要是由于在重脂质II色谱图中比在重脂质I色谱图中更大的两个尖峰。它们在VVA+范围内,因此它们没有被赋予单独的保留时间,但是它们是在大约19.85和20.15分钟,胆固醇在18.97分钟。这两个扫描显示如下。20.15分钟峰与胆固醇的比率对于两个级分随着反应时间/重脂质转化都增加。在级分I中,其从大约33%开始和增加至大约50%。在级分II中,其从大约66%开始和增加至100%,即1:1。这些可能是其他甾醇类,其中一些最初在海藻油中游离,如胆固醇,但是其中一些可能作为甾醇酯存在。随着反应进行,这些被转化为它们的游离甾醇形式和最初与甾醇酯化的相应的FFA的乙酯。这最有可能是该甾醇与胆固醇的比率如何随时间而增加。由于它们具有比胆固醇更长的保留时间,所以可能它们具有更高的分子量。它们可以是胆固醇的氧化形式。本实验清楚地告诉我们,“污泥”含有大量的官能化的海藻FFA’s,包括EPA。仅仅为了更容易地处理更流动的乙醇-可溶性海藻油而将其去除是不明智的。海藻油应当被“完全”处理。这通过以下显示:随着由于它们反应成为乙酯和FFA’s而重脂质峰减小,C16和EPA物质的总面积百分比逐步增加。在实验后测量两个级分的pH。较轻级分是pH=1.11而较重级分是pH=0.67。两个反应似乎反应至相同程度,因此该pH差异(都为非常酸性的)似乎没有造成反应性的差异。然而,测量是不同的,可以表明在较轻、更可溶的级分中存在更“酸中和性的”组分。在反应完成后,合并两个级分并且将其31.34g离心。收集25.70g的较轻层和5.63g的较稠底层。然后用等重量的无水乙醇洗涤/重悬浮底层,并且再离心,再次产生较稀顶层和较稠底层。倾析顶层并加入另一等分试样的乙醇,再次重复该过程。在三次这样的乙醇洗涤、离心和倾析后,在最后的离心管中留下4.46g的乙醇-湿固体。离心管中的固体形成三个明显的有色层。顶层是粘稠黑色,中间层是灰色,底层也是黑色但是比顶层显著更浅。从它们的相对体积判断,这三层的量大约为3:1:0.5。风干仅留下~2g的固体。那代表大约6.5%的所用的原始反应混合物,但是从31g的初始反应混合物中离心的几乎35%的重残余物(5.63g)。我们本可以进一步研究海藻油直接酸催化转化为其乙酯,但是大约同时,使用直接酸催化转化的中试工厂中的脂质转化被认为是不充分的,因此我们放弃关于该方法的工作,而是重新将我们的努力集中在之前对于FAME工艺限定的多步方法。然而,考虑仍在贮存时乙醇萃取的海藻油的“预反应”可以仍然是有益的。酸预处理证明是有效的但是不足的/不充分的。另一方面,用强碱预处理应当起作用,并且由于我们返回至那个,由于我们返回至那个方法,对于我们方法的第一步,可能值得考虑。部分II.海藻油的碱催化的酯交换/皂化乙醇的使用为工艺带来几个新的挑战。尽管乙醇更好地工作来从藻类回收所需重脂质,但是其也萃取其他极性组分,所述其他极性组分又对工艺提出新挑战。水是主要的新污染物,其完全改变了工艺第一步的方向。极性乙醇还从藻类中萃取其他极性未知物,未知物似乎增加讨厌的固体和半固体(污泥),其在工艺中显现它们自身。这要求插入另外的萃取步骤到工艺中。由于乙醇/萃取的海藻油导致对皂化的酯交换变化:FAME’s(甲酯工艺)工艺的第一步是海藻油与甲醇和甲醇钠反应,将脂质转化为游离脂肪酸的甲酯和钠盐的混合物(Na+皂)。这是典型的碱催化酯交换化学,其在生物柴油化学中常规实践,当水和FFA’s适度低时其工作良好。太多的水或FFA’s制造太多的皂,其严重损害工艺的可操作性。酯与Na+皂的该比率还取决于进料中最初存在多少水和FFA。使用己烷萃取的海藻油,残余水的浓度低(~1%),因此甲酯与Na+皂的比率高,约10/1。然而,乙醇萃取导致比己烷萃取多得多的残余水(4-6%),这么多的水将乙酯与K+皂的比率翻转至大约1/10。我们还从使用甲醇钠的商购溶液转换为通过组合KOH和乙醇制备的乙醇钾。乙醇钾本身是非常昂贵的并且难以以商业量获得。然而,从KOH和乙醇制备乙醇钾制造甚至更多的水。进行从钠到钾的转换是因为,根据生物柴油的经验,K+皂比Na+皂更容易处理。另外,K+盐具有作为肥料的潜在的副产物价值,而Na+盐有处置问题/成本。由于现在存在的水的量导致如此多K+皂的产生,所以我们最终决定放弃制备我们自己的乙醇钾,而简单地使用KOH水溶液(50重量%)将所有可皂化物转变为K+皂。如果存在足够的乙醇帮助溶解海藻油,则该反应工作良好,并且发现脂质转化在80℃小于71/2分钟内完成,只要存在足够的强碱即可。对于该皂化,推荐在海藻油中最少15-20%残余乙醇。实验室研究显示,15重量%KOH水溶液并不是非常充足将所有脂质转化为K+皂。增加至18.5%的KOH水溶液似乎对于所用的海藻油样品是足够的,但是我们后来发现,20%KOH水溶液是更好的选择。该量涵盖了我们遇到的藻类组成的各种变化。似乎海藻油的分层(如从萃取/汽提中得到)可以在贮存期间发生。推荐将该物质保持温热并且充分搅拌以避免该潜在复杂情况。实验、结果和结论:最初范围-发现使用KOH/EtOH的运行-模拟FAME工艺:在转换到我们当前的工艺(使用KOH水溶液将所有脂质转化为K+皂)之前,我们的确尝试模拟原始FAME工艺。因此,我们首先确定该新的乙醇萃取的海藻油将需要多少强碱来实现总脂质转化(使用无水乙醇中的KOH)。在这些条件下,乙酯和K+皂形式的混合物以取决于存在多少水的比率形成。假定时间足够,存在于这个系统中水不可逆地导致化学计量量的皂的形成。将5g海藻油样品和乙醇合并并与3g的25%重量%KOH在无水乙醇中的溶液混合。之前在FAME’s系统中的工作已经教导我们,这些反应是快速的,因此我们评价在100℃在Fisher-Porter管中转化的程度。我们尝试以3,8和15分钟间隔分析反应,但是很快认识到在反应时间测量中却存在如此多的不确定性,因为反应实际上在从室温在100℃的加热时间期间(预计在5分钟)开始并且即使在冷却回到室温期间也持续(如果还未完成)。因此,尽管我们不能准确地获得关于该反应的速率数据,但是我们仍然能够确定实现总脂质转化的大致强碱要求和对于该完全转化的最少时间。通过在100℃在3,8和23分钟的反应时间后(接着在再浸入油浴中后5分钟的加热/再加热时间)将反应器管冷却获得样品,并且取出1g样品用于后处理和GC分析。每个样品与0.25g的硫酸,5ml水和5g己烷混合。在剧烈振荡和20分钟离心后,将一部分顶部己烷层使用衍生化方法通过柱上GC分析。我们寻找色谱图的“重脂质”部分(19.5min至22.5min)中的峰的“消失”和FFA/FFA乙酯的比率。我们还寻找“总峰面积”,因为非常精确和准确地选取和称重每个样品。首先三个GC几乎相同,基本上可重叠。所有加入的强碱消失,并且仍剩余未反应的重脂质。显然,我们需要额外的强碱来完成反应。加入另外的无水乙醇中的3.02g的25%KOH,重复反应工序。下表显示在第一次和第二次加入KOH/EtOH后的反应结果。在100℃的海藻油和KOH/EtOH的反应。*乙酯和FFA面积%的总和;**在第二次加入KOH/EtOH后的结果。随着第二次加入KOH/EtOH,发生另外的酯交换/皂化。GC的“重脂质”面积%立即下降至稳定的4.7-4.9面积%。相应地,C16物质(酯+FFA’s)的面积%和EPA物质(酯+EPA)的面积%两者都增加并且保持恒定。已经消耗了所有重脂质。然而,酯与游离脂肪酸的比率,在每个情形中,在温度下随着时间增加而继续变化。FFA浓度在以它们的酯为代价的情况下增加。该观察结果,以及重脂质中或C16’s和EPA的总面积%中没有随时间进一步改变,表明在第二次加入后现在存在足够的强碱来实现总脂质转化。随着酯仍经历皂化,酯与酸的比率继续改变,因为仍然存在残余的强碱和水。这还显示碱催化的重脂质向它们的乙酯的酯交换要快于随后的那些乙酯向皂的皂化。该研究结论是:当反应在乙醇中进行时,通过将其与15%KOH反应,乙醇萃取的海藻油可以被转化为其乙酯和K+的混合物。然而,存在的水导致K+皂相对于乙酯占优势,为大约3或4:1。数据显示,在100℃在8-10分钟下脂质转化是完全的。这来自5分钟加热期,3分钟运行时间和1-2分钟的冷却时间的总和。随后,我们将确定,当完全皂化使用KOH水溶液进行时,该反应在80℃少于71/2分钟内完成。因此,这两个发现是一致的。尝试直接酯化酯交换-皂化的海藻油:在确定我们是否可以直接中和并酯化该碱处理的海藻油的尝试中,如我们之前在FAME工艺期间已经实现的,我们使用稍少的KOH将另一批次的乙醇萃取的海藻油与KOH/EtOH反应,因为之前的测试已经告诉我们15%是足够多的。我们将50g的海藻油和50g的无水乙醇与在无水乙醇中的22.5g的25%KOH(11.25%KOH,相对于海藻油)合并,并且将其在100℃反应15分钟。根据标准步骤准备样品用于GC分析,结果显示,11.25%KOH不完全足以转化所有脂质。总脂质面积%仍然是16.66%,显著高于使用15%KOH运行获得的4.7-4.9%,认为其是“完全的”。然而,根据FAME工艺,我们还知道,一些残余脂质可以在酯化期间转化为它们的甲酯。因此,我们决定“原样”使用该反应混合物,并且跟进该少量的残余脂质,看看它们是否同样将在FAEE酯化步骤期间转化。记得,即使在FAME工艺开始,也需要过量酸用于酯化。在乙酯工艺中使用这种新的乙醇萃取的海藻油或多或少需要过量的酸吗?将117.74g的KOH-处理的海藻油反应混合物转移至圆底烧瓶中并用13.81g的50/50硫酸/乙醇处理。这是中和之前反应中使用的所有强碱(0.10当量的强碱)所需的计算量的酸(4.9g=0.10当量的酸)加上足够的额外酸(2.0g=大约4%相对于原始海藻油重量)以使得反应混合物呈酸性。将反应混合物回流(94-96℃)1小时,每20分钟去除样品在通常样品后处理后用于GC分析。第一轮分析显示无反应,所以加入另外的硫酸并且在取样的同时继续反应另外1小时。在酯化超过90%酯化之前需要几次额外加入的酸。结果总结在下表中。在“回流”(94-96℃)下的碱处理(KOH/EtOH)的酯化-第一次酸运行*乙酯和FFA面积%的总和。显然,在该第一硫酸浓度下很少反应发生。酸已经被海藻油中的碱性组分总和。在“时间零点”样品和样品的其余部分之间存在轻微的变化。也许酸在被中和前短时间有效。加入另外1.03g的浓硫酸,并且重复实验。结果如下。在“回流”(94-96℃)下的碱处理(KOH/EtOH)的酯化-第二次酸运行*乙酯和FFA面积%的总和。再次,似乎没有反应发生,因此加入另外1.04g的酸,再次重复工序。结果如下。在“回流”(94-96℃)下的碱处理(KOH/EtOH)的酯化-第三次酸运行*乙酯和FFA面积%的总和。现在我们开始看到,C16FFA’s和EPA两者的百分比酯化随时间轻微的和非常缓慢的增加。我们现在具有足够的酸存在以刚好克服海藻油中的碱。我们使用50g的乙醇萃取的海藻油开始该反应。因此刚好花费8%酸(相对于该海藻油)来中和碱。并且该海藻油仍然是20%乙醇。这比使用己烷/MeOH萃取的海藻油的FAME’s工艺中所需的是要高得多的酸要求(~3%)。更极性的乙醇(加上比己烷更高的回流温度)明显比己烷/甲醇从海藻中去除更多的碱性物质。这仍然是太少的酸而不能催化酯化,我们看到残余重脂质没有变化,因此加入另外的酸。此时我们加入2.01g酸并且重复小时回流期。那些结果如下。在“回流”(94-96℃)下的碱处理(KOH/EtOH)的酯化-第四次酸运行*乙酯和FFA面积%的总和。酯化现在很好进行,但是我们还未转化任何大量的残余重脂质。另外,酯化百分比仍然不令人满意(97.5%最低值)。%酸现在是12%,远远大于FAME工艺和己烷/MeOH萃取的海藻油曾经需要的。为了看看是否甚至更多的酸将开启残余重脂质的转化,再加入一次酸(1.0g使得酸浓度达到14.2%!)。那些结果如下。在“回流”(94-96℃)下的碱处理(KOH/EtOH)的酯化-第五次酸运行*乙酯和FFA面积%的总和。现在,在14%酸(相对于初始加入的海藻油)下,酯化已经达到平衡,残余的重脂质慢慢被转化为另外的乙酯。这在对于C16和EPA两者的增加的面积%方面是明显的。然而,这似乎使用多得多的硫酸,并且,更重要的,对于允许达到目标&酯化目标来说似乎存在太多的水。这对于己烷/MeOH萃取的海藻油(其中存在仅1%的残余油)并非如此。使用该进料,甚至乙酯可以完成酯化达到或接近97.5%。先前关于生产FAME’s的工艺似乎不能对于FAEE’s起作用。我们需要一种新方法,其可以为我们提供“干燥的”FAA’s用于酯化。并且这就是我们下一步的开发。接下来描述该工艺。新型乙酯工艺概念化:新型工艺概念是完成含乙醇的海藻油的完全水溶液皂化。这将所有脂质转变为K+皂。该皂然后将通过用10%硫酸水溶液中和转变为它们的FFA形式。接下来,FFA’s将被萃取到适当溶剂中,产生FFA’s在烃中的溶液。己烷,环己烷和庚烷最初被认为是适当的萃取候选物。在FAME’s工艺期间,我们已经认识到可以使用含有高达50%己烷的FFA溶液进行酯化,因此我们有少许自信该方法也将在此起作用。在酯化后,利用水的第二次萃取将去除酸和乙醇,并且得到产物乙酯在烃中的溶液。烃的蒸发将得到乙酯加上其他非极性不可皂化物的混合物,其用于进一步分馏。水溶液皂化-实验、结果和结论:中试工厂的海藻油进料已经稀释至50%乙醇以易于处理。将该物质的样品用于以下实验,但是其通过在旋转蒸发器上加热至50-60℃、保持28英寸压力直至去除计算量的浓缩物而首先浓缩为25%总挥发物的目标。总挥发物分析显示其为22.2%总挥发物。将少量的额外乙醇加回至用于下列实验的样品,以使所用的所有“海藻油”样品含有25.0重量%总挥发物(乙醇加上水)。为了确定对于总脂质水解需要多少KOH水溶液(50%水溶液),将样品与3种不同量的KOH水溶液(相对于所用海藻油的重量,15和21%KOH之间)反应。将每个样品保持在78-82℃于密封离心管中达1小时,整个运行过程中间歇振荡。样品充分液化,并且似乎均匀。在位于水浴之上的管的上部中可以看到乙醇的回流/冷凝。0.5G样品用1g的10%硫酸水溶液和5g的己烷处理。在剧烈振荡后,接着进行15分钟离心,产生不含酸和乙醇的顶部己烷层。这些通过柱上衍生化GC来分析,以获得下列数据。在78-82℃使用50%KOH/H2O的海藻油的皂化。*Wt%KOH是相对于所用海藻油的重量。**VVA+是RT在19.5和22.5分钟之间出现的重脂质的总面积%。相对于海藻油,18.37重量%KOH可能足以将脂质和乙酯皂化为K+皂,但是在20.35%KOH下似乎还有另外的脂质转化发生。这里,VVA+峰降低至4.70面积%(我们较早对于使用KOH/EtOH的“完全”转化看到的值)并且胆固醇值增加至6.17%。甾醇酯,如胆固醇酯(其已知是通过Nannochloropsis制成),很可能是最慢皂化的重脂质,这是因为酯官能团周围的空间位阻。因此,该观察似乎在化学上有道理。如果有大量的EPA被胆固醇酯化,则使用20%KOH目标用于该水溶液皂化反应可能更好。使用50%KOH水溶液的皂化的最少转化时间:当该下一个实验开始时,还未获得前一个GC数据。如果它们已经获得,我们将利用20%KOH。相反,这些“速率”实验使用18.48%-18.81%KOH进行。在并行实验中在浸入82℃水浴的密封离心管中,海藻油样品与大约18.5%KOH水溶液(50重量%)加热7.5,15和30分钟。在流动的冷自来水冷却后,通过用1g的10%硫酸水溶液处理,照常制备单个的0.5g样品。充分振荡样品,各自与5g己烷等分试样合并。在振荡和离心后,顶部己烷层是用于衍生化柱上GC的样品。结果如下。在78-82℃使用50%KOH/H2O的海藻油的皂化。*Wt%KOH是相对于所用海藻油的重量。**VVA+是RT在19.5和22.5分钟之间出现的重脂质的总面积%。基于这些数据,皂化在甚至最短的反应时间内出现。另外,三次GC扫描似乎几乎可重叠。在该KOH浓度下,VVA+和胆固醇值可与先前1小时运行相比。将这三个运行与先前1小时运行再次比较,表明20%KOH是对于强碱更好的目标。将此推荐给中试工厂他们使用。中和与萃取:在将海藻油完全皂化为游离脂肪酸(FFA’s)的K+皂后,下一步是用硫酸水溶液酸化,将所有皂转变为FFA’s。这允许它们从酸性水中回收,并且所有海藻油副产物通过萃取仍然溶解在水相中。通过加入10重量%硫酸水溶液进行中和,直至达到大约1.5的pH。理论上,在pH为3左右所有皂将转化回为它们的FFA形式,但是似乎在较低pH下相分离更好。这后来在中试工厂中证实。在研究的这个部分期间,我们还认识到己烷和庚烷是用于回收FFA’s的良好萃取溶剂,但是环己烷不是。环己烷具有比己烷(0.66)或庚烷(0.68)更高的比重(0.78)。这使得其从水层的相分离更慢和更难。在实验室萃取研究期间,我们还认识到在50℃的萃取比在25℃的萃取更快和更完全。如果对于萃取在中试工厂中制备的更充分混合的中和混合物而言实际上不需要,则中试工厂的后期工作显示60-65℃甚至更好。可行的是,65℃萃取可以与环己烷一起工作,但是一旦认识到庚烷的优势,没有研究这个。在室温使用己烷或庚烷,需要3个分批萃取,使用等重量的溶剂以获得皂化反应混合物的中和/酸化产生的FFA’s的98%回收。实验室中的相分离室温下花费大约30分钟。然而,环己烷更加困难。相分离花费时间更长,并且需要至少4个分批萃取来实现可与己烷或庚烷相比的回收。使用我们的小型Scheibel柱在实验室中进行了多个连续萃取研究,但是那些结果没有包括在本报告中。我们尝试在海藻油从其最初状态通过皂化、中和/酸化、萃取和酯化的转化全过程之后接着用十八烷掺杂原始海藻油。我们希望在十八烷之后接着进行GC。不幸地,该努力是不成功的,因为几乎不可能准确定量原始油或沿着工艺路径的十八烷。原始油中的乙醇和水、来自溶液的不溶物和来自工艺期间溶剂的稀释的存在使得封闭任何合理的物质平衡几乎不可能。部分III.烃溶剂中的酯化研究新“假想的”FAEE工艺要求中和的皂化反应混合物的烃萃取。预期这产生15重量%FAA’s的相对干燥的烃溶液。FFA’s在该溶液中然后被酯化为它们的乙酯。从产品注册观点来看,Aurora’s有理由期望使用环己烷而非己烷将是有利的,因此我们首先开始确定在环己烷溶剂中FFA将酯化成什么样子。根据该新工艺的萃取步骤预期FFA’s在环己烷中的15重量%溶液,因此我们计划我们的实验使用该浓度。通过从刚才描述的KOH水溶液皂化反应混合物中回收FFA’s,我们获得了我们的原材料。皂化反应混合物全部合并和用10%硫酸水溶液酸化,将所有皂转化为FFA’s。它们然后被萃取到己烷中,将振荡的混合物使用离心分成两相。己烷的蒸发得到8.30g的FFA’s加上其他可萃取的不可皂化物胆固醇,烃等)和残余重脂质。向该混合物加入47.04g的干燥环己烷,得到15重量%“FFA’s”在环己烷中的混合物。环己烷中的该储备溶液用于所有随后的环己烷实验。开发“基本情形(basecase)”步骤用于进行(我们将在许多不同条件下进行的)多个酯化。这里详细描述该一般步骤。除非另外指出,在所有情形中遵照该步骤。用于酯化实验的一般酯化步骤:将5.0g的储备FFA溶液加入50ml离心管。加入目标量的乙醇,接着加入指定量的浓硫酸。所有乙醇(无水,除非另外指出)和所有硫酸来自相同来源。将C管密封和放置在温度受控的水浴(保持在被研究的指定温度下)中。在运行指定时间后去除C管并且通过将它们置于流动的冷自来水下达大约2-3分钟而冷却。然后遵循两步之一。如果运行完成,则将C管离心15分钟,此刻之后去除顶部烃样品并提交GC分析。如果继续运行另外的时间,去除大约1/3的充分振荡的样品并且如刚才所述离心/取样。然后将剩余的2/3的样品重新密封并且返回至水浴中达其另外的反应时间。然后如前将其离心和取样。对于这些酯化研究,我们需要比柱上GC方法已经提供的更好的分析步骤。通过使用DB-1柱和分流注入,实现了EPA乙酯和EPA的TMS酯的基线分离。对于本报告的该部分描述的所有酯化,使用该方法来确定酯化程度。对于这些酯化实验,仅提供%EPA酯化。照常,通过用EPA乙酯的面积%除以EPA乙酯的面积%和其EPA的TMS酯的总和并且用100%乘以该商,得到%酯化(或乙酯选择性)。很明显,可以从任何一对的FFA乙酯和它们的TMS酯计算乙酯选择性。在环己烷溶剂中的酯化结果:在下表中提供了在环己烷溶剂中进行的酯化的结果。在75℃在环己烷中15%FFA’s的酯化。运行#%H2SO4%EtOH℃运行时间(min)%EPA酯化6-1215753095.07-2210753082.47-3220753094.37-43*16753071.28-5215(共沸)**753059.28-6115753088.38-7315753086.69-1215754593.99-2210754591.1*仅使用环己烷中的3%FFA溶液。任何其他事物均与存在的海藻油成比例。**共沸”乙醇是通过加入95%总量的乙醇和5%的水制备。这些酯化结果最初是令人失望的。我们在这些运行期间没有实现最低目标酯化结果(97.5%),但是通过重复我们最先的两个运行达更长的时间,我们确定我们在仅仅30分钟内没有达到平衡(参见7-2相对于9-2)。因此,在环己烷中的这些运行中的大部分反映了反应速率差异。酯化似乎是均匀的或至少是充分乳化的。在反应后离心后明显存在两个相,但是相分离并不如在FAME工艺中使用存在的己烷先前观察到的那么快。在离心后观察到的重相似乎非常稠并且是污泥状的。开发校正曲线用于分流注入GC方法,使用月桂酸甲酯(ML)作为内标。使用ML添加,我们证实我们的储备溶液的确如所预期的为15重量%。使用内标,GC6-1显示存在13.9%总乙酯和它们相应的FFA’s。此外,样品中存在1.3%胆固醇。因此,总脂质分析是15.2%。我们预期按重量计15.0%。此外,乙酯含量为4.95wt%;EPA为另外0.25%。因此,总“EPA”含量是5.2重量%。这是“良好的”海藻油。该系列中的酯化运行之一利用环己烷中的仅3%FFA’s溶液。所有其他试剂也按比例缩小20%。该结果可能是由于较高稀释度导致,因为酯化是双分子反应。对于这些条件本应当使用更长的反应时间。一个酯化尝试使用共沸乙醇作为反应物。该运行中存在的多余的水极大地减慢了速率。基于证明我们在仅30分钟内没有达到平衡的其他运行(7-2相对于9-2),该运行也本应当运行更长以确定在这多余得多的水存在下什么样的最终酯/FFA平衡是可能的。同样,当其终止时,我们仅使用1%酸进行的该运行没有达到平衡。未来实验中将进行更长的运行。在实验室中进行几个批次的酸化皂化反应混合物的环己烷萃取。发现萃取相当困难。相分离需要长时间,需要多个萃取来回收所有的所含FFA’s。在50℃萃取得到的结果比室温要好得多。由于使用环己烷的萃取挑战和我们差的最初酯化结果,我们返回使用己烷用于萃取和酯化。使用大豆油的对照酯化-重要的观察/结论:为了独立评估在该反应系统中我们可以期望多少酸催化的酯交换,我们将大豆油样品(99+%甘油三酯)经历运行7-1中所用的相同条件。这将为我们提供关于以下的更好理解:在酯化期间我们可预期多少残余重脂质转化。大豆油是几乎纯的甘油三酯,我们计划通过GC确定转化,正如我们对于FFA酯化那样。将0.75g的无水乙醇加入0.75g大豆油在4.25g的环己烷中的溶液。该溶液是均匀的和完全透明的。然后,加入0.10g的浓硫酸,并且振荡混合物。该溶液变得朦胧并且形成少的第二相。这是硫酸,其在环己烷/大豆油相中的溶解度已经被超出。这显然证明,当反应在烃溶剂中进行时,在酯化过程中将始终存在两相。该第二较重相的体积明显比加入的硫酸的量大。一些乙醇被拉出油相以将硫酸溶剂化。相应地,我们预期一些硫酸也分配到油相中,因为残余的乙醇仍然存在于该相中。这在酯化发生的情况下是可能的。该观察中有趣的可能性是在酯化期间形成的水应当快速超过其在油相中的固有溶解度和被吸入重硫酸相中,在那它比乙醇更好地将硫酸溶剂化。这是允许酯化进行的程度比如果反应在单一均相中发生更深的机理。在30分钟后,冷却反应混合物,离心并取样用于衍生化GC分析。由于预期甘油三酯在产物混合物中,该分析利用柱上方法。GC6-C(对照)表明大部分(~60%)的大豆油甘油三酯保持未改变,并且反应的量仅仅进行至二甘油酯阶段。仅大约1-1.5%的油酸乙酯和棕榈酰油酸乙酯被检测到,连同大约0.9%的油酸单甘油酯。这些物质是预期的产物,因为大豆油非常富含油酸甘油酯。该对照实验的结果与在直接酸催化的酯交换研究期间进行的发现非常一致。在这些条件下,甘油酯仅以非常慢的速率被转化为它们的乙酯。重要的是记住在这些酯化过程中清楚观察到两相。已经一直很明显在FFA酯化结束时存在第二重相。但是那些溶液/反应混合物非常深,并且还未确定减少的重相是不同于海藻污泥的任何事物。本实验证明了存在的过量酸分成该第二重相并且其吸引乙醇和非常可能水至其自身中。关于酯化的另外的FFA的制备:在这些最先酯化运行期间,环己烷中的15%FFA溶液被消耗,因此此时在己烷中制备了新溶液。其通过将2.3g的乙醇加至具有19.2%总挥发物的27.08g的海藻油来制备。这产生29.38g的海藻油,含有25%总挥发物。加入5.2g的KOH(10.4g的50%水溶液),将混合物在705C加热1.5小时。然后,加入60g的10%硫酸,将获得的振荡混合物与~100g的己烷混合,再次振荡并离心。将己烷相分离并在旋转蒸发器上蒸发至恒定重量,产生12.71g的残余油。将这放回至72.20g的己烷中以制备“FFA’s”在己烷中的15重量%溶液,用于下一组实验。在己烷溶剂中进行的酯化的结果在下表中提供。在75℃在己烷中15%FFA’s的酯化。*使用己烷中的仅3%FFA溶液。任何其他事物均与存在的海藻油成比例。**共沸”乙醇是通过使用95%目标重量的乙醇和5%的水制备。***这些运行的后处理包括添加3g的水于反应混合物或样品中,并在离心前将其充分振荡。这是通过酸催化的水解来评估逆转(reversion)。来自环己烷中酯化的发现帮助我们在己烷中运行更好的实验。我们知道运行需要更多时间来达到平衡,因此大多数我们在己烷中的运行在30分钟和60分钟都取样。我们在己烷中的第一次“基本情形”运行的结果令人困惑。6-1和12-1在相同条件下运行,但是己烷运行比其在环己烷中的对应物的转化要少得多。于是我们认识到,尽管目标为75℃,但是己烷运行可能在较低温度下进行。在这些运行中,观察到己烷在水浴上C管的冷却器壁上回流和再冷凝。己烷在680C沸腾;环己烷在81℃沸腾。较低温度似乎最好地解释为何己烷运行比环己烷运行更慢。不管怀疑的低温,其他己烷相对于环己烷比较显示在可比较的反应时间,相比于环己烷,对于己烷的更好的结果。参见7-4相对于13-3*(3%FFA浓度)和8-5相对于14-2(使用共沸乙醇)。回流可能实际上导致己烷运行更好的连续混合,并且混合可能对于存在两相的反应是重要的(如果在游戏中存在重要的物质转移因素)。当它们的浓度随着酯化程度而改变时,酸,水和乙醇可以在相之间移动。如果两相保持充分分散和彼此高表面积接触,则将促进这种转运。预期从油相中去除水是酯化的驱动力。此刻,这些思想仅仅是推测,并且如果重要的话,可能使得有理由未来进行进一步的研究。总之,由于许多原因,己烷结果是非常有希望的。在许多运行中观察到了大于97.5%的酯化。使用仅3%FFA溶液的酯化在30分钟后达到95.2%酯化和在60分钟后达到98.8%。使用共沸乙醇,使用高酸浓度(3.4%)的酯化,在30分钟后达到96.6%和在60分钟后达到97.6%。如所预期的,酯化的酯化速率和程度由高酸浓度驱动。尽管在大部分的60分钟运行中似乎已经达到平衡,但是可能需要另外的时间再进一步改善向乙酯的转化。在该系列中运行两个实验,以确定在酯化后在水萃取期间“逆转”是否将是一个显著问题。该萃取是预期的,因为它是从烃溶剂中的乙酯中在将它们干燥和分馏之前去除残余乙醇和酸的有效方式。因此,改变对于运行14-2***和14-3***的后处理以研究该可能性。按照通常的后处理(顶部烃层的离心,取样等)分析14-2(60分钟运行)和14-3(30分钟运行)。然而,它们的重复,14-2***和14-3,***,在冷却后但是在离心前,向反应混合物中加入3.0g的另外的水。在加入该另外的水后,将反应混合物充分振荡并然后照常离心,取样和分析。两个比较都表明一些“逆转”(水将酯/FFA平衡往回推向错误方向)。运行14-2(非水性后处理)分析为97.7%酯化,而其水后处理对应物14-3,***显示93.6%酯化。这是4.1百分点的差异,并且如果不能避免将产生严重问题。14-3相对于14-3***也显示%酯化的下降(98.2%相对于97.0),但没那么严重,仅损失1.2%酯化。较大的差异可能是因为存在于运行14-2(其与共沸乙醇制备)中的更大总量的水。14-3的运行是使用无水乙醇。在水加入后的额外搅拌(振荡)可能导致两相之间更多的表面积接触,由此促进反向水解反应。再次,这些是需要更多检验的假说。有充足理由说,在水洗涤后在水洗涤反应混合物期间可以预期一些逆转。然而,注意这些结果是使用利用3.4%的高酸浓度的运行获得的。逆转是酯化的逆转并且也是酸催化反应。在酯化期间的较少的催化剂,即较低酸浓度,应当降低该反应速率。另外,如果重酸相从反应混合物中在进入萃取柱之前通过倾析或离心分离,则应当完全避免逆转。实际保留在酯溶液中的酸的量应当快速转移至萃取水中,此时甚至逆转的可能性应当终止。作为避免逆转的措施,强烈推荐在水洗之前倾析。作为旁白,如果我们必须在升温下运行萃取柱来促进相分离,则倾析掉酸首先变得甚至更重要。如其对于所有化学反应,高温将驱动更快的逆转。在己烷中的这些酯化期间发生的一个另外的观察值得关注。运行13-2(60分钟运行)的GC分析显示几乎不存在胆固醇。实际上,峰在较早的保留时间出现,其鉴定为3,5-胆甾二烯,胆固醇的脱水产物。在化学上,脱水讲得通,因为这是使用额外的酸3%进行的运行。但是在环己烷中运行8.7(3%酸)具有正常的胆固醇浓度。进一步混淆该情形,在30和60分钟两者的运行14-2,***显示大大降低的胆固醇浓度,运行14-1(13-2的重复)在GC扫描中在30分钟后显示几乎无胆固醇,但是在60分钟的扫描显示大约2/3正常量。此时,对于该观察的清楚解释仍然难以捕获。然而,我们还看到一些Aurora样品的GC扫描中胆固醇的损失。在那些情形中,胆固醇损失始终与极低pH相关。高酸强度也存在于这些己烷运行中,所述己烷运行显示胆固醇损失但是不容易解释的是胆固醇如何能够在30分钟消失和在60分钟重新出现,除非酸度在酯化早期当存在较少水时更强。随着酯化进行和产生更多的水,酸强度将降低,并且也许,在较低pH形成的3,5-胆甾二烯,随着pH增加再水合为胆固醇。如果该问题变得重要,则可以在独立研究中进一步研究。表明庚烷可能成为从各种进料中分离油的萃取工艺的选择溶剂。庚烷(b.p=98-990C)的沸点比己烷的沸点(b.p.=68-69℃)高大约300C。然而,庚烷和己烷在25℃具有可比较的比重,庚烷为0.681和己烷为0.657。这意味着它们应当具有类似的萃取特性,但是庚烷可以在比己烷更高的温度下使用而不超过大气压。环己烷沸点(81℃)适中是可接受的,但是具有高得多的比重0.779(20℃)。其比重接近水,这最可能导致其比己烷或庚烷更差的萃取性质。它正是需要更长时间从水溶液进行相分离。以这个观点,我们进一步评价了在庚烷溶液中并且利用其沸点在高温下的酯化。下表总结我们在90℃的结果。通过将用于之前酯化的FFA’s的剩余己烷溶液汽提至恒定重量,制备在庚烷中的15重量%溶液的FFA。将6.86克残余油重新溶解在38.87g的庚烷中,以得到在庚烷中的15.0重量%的FFA。与较早的实验一样,该溶液的等分试样用于在庚烷中进行的所有酯化。在制备FFA’s的庚烷溶液后,该溶液置于实验台顶部过周末。周一早晨,在瓶子底部观察到大量的污泥。因此,将瓶子充分振荡,并取样用于GC分析。将另一个较少样品离心,沉积少量的污泥于C管底部。将离心溶液取样也用于GC分析。该试验的目的是确定污泥是否“含有”大量的FFA’s。使用ML内标方法和两个SC扫描的面积计算,GC分析总体表明离心溶液含有在未离心样品中发现的FFA’s的仅大约81%。这表明沉淀的污泥吸收/吸附一些FFA’s。认为这只是一个物理现象,因此将溶液留下未离心并且在取出样品酯化之前始终重新振荡。在庚烷溶剂中进行的酯化的结果提供在下表中。在90℃在庚烷中15%FFA’s的酯化。*使用在庚烷中的仅3%FFA的溶液。任何其他事物均与存在的海藻油成比例。**使用十二烷基苯磺酸作为催化剂(均匀溶液)。数据检查告诉我们,在90℃在庚烷中的酯化是成功的。几乎每次运行超过97.5%酯化选择性目标。虽然对于30至60分钟之间的大多数运行选择性持续提高,但是仅在该30分钟时间点之后反应已等于或高于目标选择性。显然,该额外的温度促进酯化比在75℃显著更快地完成。一些通用结论包括:(1)仅使用10%无水乙醇,在90℃在仅30分钟之后,可以仅使用1.2重量%的硫酸获得98.2%的酯化选择性。在接下来的30分钟内选择性确实提高至99.1%(19-1和19-2)。如果在这些相同条件下使用共沸乙醇,对于30和60分钟的反应时间,选择性分别下降至79.2%和89.2%。这可能是89.2%还没有处于平衡。更长的时间可以实现更大的选择性。(2)然而,利用相同的10%共沸乙醇,将酸浓度增大至2%在60分钟之后足以达到98.3%的酯选择性。由于存在的额外的水的减速作用,在该反应的前30分钟之后选择性仅达到87.4%。这是显著的结果。据信使用共沸乙醇达到此高酯选择性的能力是有可能的,因为存在两个分离的相。将水从反应烃相吸出进入到重的含硫酸极性相中,在那里它被占用(tiedup)使浓硫酸水合/离子化并且被阻止进入到化学平衡中,如同如果它是游离水时那样。如果存在一个相,则将不会达到这种酯化程度。在类似反应条件下用十二烷基苯磺酸代替硫酸在30分钟之后得到大约相同的酯选择性(82.0%),如在30分钟之后在16-5中观察到的。在这种情况下,所有磺酸保持存在于反应相中。据信其浓度接近于仍然溶解于烃相中的硫酸的浓度。(3)令人惊奇地,使用10%共沸乙醇,当在30分钟时间点测量时,反应在3%硫酸存在下比在2%的情况下更慢。首先令人困惑的是,我们通过另外的对照实验领会简短地描述,更多的乙醇随着添加的酸增多而从烃相除去。据信这种用于酯化反应物之一的烃相的“缺乏(starving)”导致在30分钟的降低速率。假定足够的时间达到平衡,然而,3%酸运行将使酯化达到甚至更高的酯转化率,因为它可以将更多的水从反应相拉出。在60分钟,3%酸酯化达到99.2%酯化效率,而2%运行为98.2%。尽管这些差异小,但是我们认为它们是真实的。(4)运行15-2仅采用3%FFA浓度。乙醇和酸成比例地放大。在90℃,酯选择性在30和60分钟是优越的,分别为99.2%和99.1%。这些实际上是相同的值。该额外的酸和更高的温度易于克服由于稀释所致的速率降低。使胆固醇消失和重新出现的神秘在这些在庚烷中的900C酯化的情况下继续。同样,当采用更高酸水平时出现该现象。在当使用至少2%酸时的几乎每一种情况下,即使在仍然看到胆固醇时,其浓度也从正常5.3-5.5%降低至4%以下。为了更好理解此现象,我们利用溶解在具有20%乙醇和3%硫酸的庚烷中的可靠胆固醇(36.2mg)和胆甾烷(5.1mg)(胆固醇的-OH被-H替代)运行实验。这基本上运行15-2达60分钟,同时没有海藻油FFA’s存在。GC16-2仅显示4个峰,一个用于胆固醇,在25.559min.(16.9%),一个用于胆甾烷,在23.831min.(8.70%)和两个新的峰。这些峰之一刚好在胆甾烷的前面,在23.588min.(10.5%)和另一个刚好在胆固醇的前面,在25.152min。(16.9%)。这两个新的峰都在其他样品的GC扫描中看到,其中胆固醇峰“来了又去”。当胆固醇经历脱水时,它形成3,5-胆甾二烯。3,-5-胆甾二烯仅比胆甾烷轻4个原子质量。它很可能是正好在胆甾烷的前面洗脱的峰。胆固醇脱水可能机械地通过烯丙基阳离子进行。此阳离子可以失去一个质子而形成3,5-胆甾二烯或与乙醇反应而形成胆固醇乙醚。在胆固醇TMS醚峰之前的峰很可能就是此乙醚。这两个提出的分子都可以转化回为胆固醇。3,5-胆甾二烯需要再添加水,反应更可能朝向运行结束,在那里存在最大量的水。同样,胆固醇乙醚可以水解回为胆固醇并且当水处于其最高浓度时,此反应同样更可能朝向运行结束发生。虽然肯定没有被证实,但是这可能是对于一些酯化期间胆固醇的来了又去的解释。存在另一种在“庚烷”表中报道为“对照”的GC,16-1。当这些新的GC峰的一些开始出现时,将新的离心管用于此系列的酯化。因此进行“无油”运行并且庚烷层经由衍生化GC分析。根本没有看到GC峰,证明从这些管中没有浸出任何物质负责该新的GC峰。此对照,加上刚刚利用胆固醇和胆甾烷描述的那个证明所述新的GC峰来源于胆固醇。支持在烃相中酯化发生的证据:烃溶剂中的海藻油FFA’s的酯化总是具有在离心后非常明显的小的、致密相。此相的存在在反应期间是不明显的,因为反应混合物是如此极黑的并且还有“不溶物质”存在,使得甚至更难以“看到”该第二相。若干“对照”实验支持/证实存在重质强酸相。这些实验在没有任何海藻油FFA’s存在下进行,所以极易可见地看到发生了什么。将庚烷(19.0ml)添加至25ml量筒。然后,添加2.0ml的无水乙醇并且所得混合物保持总体清澈。完成溶解并且该两个组分此组成下完全混溶。接下来添加0.10克的浓硫酸。将混合物振荡并且变得非常浑浊。在2-3分钟内,微小的重相分离并且在量筒的底部上是可见的。其体积看起来比添加的H2SO4的体积更大,但是其是小的并且不可能进行测量。然而,第二相明确地存在。添加第二个0.10g部分的浓硫酸,具有类似的结果。重质底层的体积明显大于所添加的0.20g硫酸的体积。最后,添加另外的0.32g的浓硫酸,用于总计为0.52g硫酸。此硫酸的重量将会仅占约0.25ml的体积。在最后的酸添加之后和在振荡并且允许相分离(1-2min.)之后,底层的体积测量为2.0ml!显然,硫酸将乙醇从庚烷“萃取”出来,在此实施例中为大约1.75ml。这是约1.75mlx0.789g/ml=1.38g或0.03摩尔的乙醇。0.52克的硫酸为0.005摩尔的H2SO4或0.010摩尔的H+。该溶剂与质子的3/1摩尔比仍然很低,不足以完全使所有硫酸水合/离子化。高度可能的是,硫酸酯化在此相中发生,形成硫酸氢乙酯和水。所得到的水分子将使硫酸氢乙酯的剩余质子水合,甚至比乙醇更好。并且硫酸氢乙酯仍然是足够强的酸来催化酯化。更早的研究硫酸和乙醇在甚至更低温度下的反应的研究支持此假设。对清澈的庚烷层进行取样并且5.21g用在甲醇中的0.01NKOH滴定,使用酚酞作为指示剂。用了2.2ml的滴定剂来达到终点。计算表明在庚烷层中存在206ppmH2SO4。所以,即使利用在单独的重质底相中存在的大多数硫酸,也有206ppm硫酸分配在庚烷/乙醇层中。这足以催化在该相中的酯化。另外,随着酯化进行并产生水,该水将超出其在庚烷/乙醇相中的溶解度并转移至重质、浓酸相,在那里它将被占用,水合硫酸/水解硫酸氢乙酯回到硫酸和乙醇。这是高度合理的。进行另一个对照实验,其进一步支持这种反应模型。在没有任何FFA’s存在下,重复酯化实验16-3。将5克庚烷、0.50g无水乙醇和0.10g浓硫酸在90℃加热30分钟。将反应混合物冷却并离心。将庚烷层离心并且样品(1.96g)用在甲醇中的0.01NKOH滴定,用了1.0ml的滴定剂来达到酚酞终点。计算表明存在249ppmH2SO4,与之前在没有使用“反应条件”的情况下进行的实验一致。我们还发现,如果我们在达到平衡之前终止酯化,对其离心并且仅对分离的烃层再加热,则容易促使酯化超过99%选择性。“其余的”酯化仅利用溶解在烃相中的乙醇和酸完成。这或许是可用于支持在烃相中发生反应的最强证据。这提议一种令人感兴趣的工艺机会。酯化可以简单地通过将剩余烃反应溶液加热另外的时间,在倾析重质、湿酸层之后“结束”。这可以是用于在进入分馏之前可能获得最高酯转化率的一个好的选择。这些对照实验进一步支持早期提议的工艺选择。在酯化后,应允许反应混合物进行相分离。轻质庚烷/乙酯层可以水洗以除去乙醇和残余酸。重质酸层可以用作中和皂化反应混合物所需的酸的一部分。支持中试工厂操作的酯化试验:中试工厂接近用于装置(单元,unit)中的酯化行动的时间。要求对提出的操作条件证实有效并且在实验室进行短暂间歇酯化研究以完成这一点。预期在己烷中的15%FFA浓度并且相对于存在的FFA,考虑了3至5%之间的硫酸的酸条件连同10%无水乙醇。实验的目的是确定在80℃在1和2小时的反应之后的酯化选择性。使用我们的在己烷中的15重量%FFA的储备溶液,我们在在Fisher-Porter管中利用10%无水乙醇运行并行酯化,在加热1和2小时之后对反应取样。使用柱上和分流注入GC方法二者,根据通常步骤(冷却、离心、顶部己烷层取样、衍生化)对样品进行处理和分析。柱上分析首先进行,但是分析的准确性不令人满意,因为EPA乙酯峰与其TMS酯的峰重叠。因此将它们在DB-1柱上重新分析,得到优异的结果。这是多个第一次之一,我们采用此方法并继续在我们需要非常准确的酯选择性测量结果的时候使用它。然而,这种方法不能显示可能存在的任何重脂质的存在,因为它们从不逃离注入口并进入柱。该简短评价的结果示于下表。在80℃在己烷中的15%FFA的酯化。第一个值来自柱上GC分析,第二个值来自分流GC分析。可以从这些数据得出若干结论。利用3%酸的一小时反应足以获得目标酯化选择性。这在使用更好GC方法时是显而易见的,因为它对于C16和EPA物质都给出酯和酸峰之间的完全基线分离。然而,如果此方法不可用,则通过仅使用C16计算可以获得指导。当使用柱上方法时,那些酯及其对应的酸比两种EPA物质更好分离。3%酸对于这种酯化可以是足够多的,因为反应在反应的第一小时内“结束”。应避免太多的酸,不仅由于经济原因而且为了使对于EPA结构中的顺式双键从顺式至反式异构化的可能性最小化。不幸地,我们当前的室内GC方法不能检测EPA的许多可能反式异构体中的任一种,但是它们是已知的并且已知这样的异构化可以通过高温单独促进或通过强酸和较低温度促进。对于此工艺,推荐使用完成此工作的最低酸浓度和使用最低的有效温度用于该工艺。确保异构化被最小化很可能是在对FAEE工艺最后确定反应条件时的决定性考虑。使用5%酸的结果不仅是不必要的并且由于可能的异构化所致而可能对EPA有害,而且相比于3%,它可能实际上更慢并且效率更低。C16数据似乎支持这一点。其他实验已显示,使用的超过实际所需的更多的硫酸通过从反应油相吸出过多乙醇而负面地影响酯化。如果我们使该相缺乏太多的乙醇(更可能的是在最小化乙醇浓度时),我们将不仅减慢速率而且甚至可能开始损害平衡的位置。像往常一样,在离心之后存在重质层。这是极性酸层。按时间顺序地,此研究在此报告的早期报道的工作之前。3%/1hr运行的速度和完整性正是导致我们尝试确定最低有效酸和乙醇浓度以及使它们有效所需的温度和时间。部分IV.分析批准了一些关于分析方法的另外的意见。藻类脂质难以使用气相色谱法准确地分析。即使使用衍生化,也有不会从柱洗脱且在GC扫描中出现的重质极性、复合脂质。甚至普通的甘油三酯也将在普通注入口中仅部分地气化,所以除非样品“柱上”注入,否则它们的定量是不可能的。并且如果人们期望看到并定量甘油单酯和甘油二酯,则衍生化也是必须的。这些都是ASTMD6584(EN14105)的一部分,所需的用于证实生物柴油的分析方法。然而,气相色谱法是用于跟进我们在此过程中运行的反应的进展和单元式操作的重要工具。我们已经利用我们在D6584和在先生物柴油工艺研究的经验并且改进这些方法以分析海藻油中的FFA’s、FAEE’s和其他组分如胆固醇、甘油单酯、甘油二酯和甘油三酯。初始地,当我们开发FAME工艺时,我们的标准GC方法确实能够区分甲基EPA和EPA的TMS-酯。这使得跟进酯化进展很容易。然而,EPA乙酯在其甲酯对应物之后洗脱并且与EPA的TMS酯重叠。没有获得这两个峰的基线分辨率,所以用于确定酯化进展或选择性的两个的积分伴有太多的误差。将分析切换到具有分流/无分流注射器以及在EPA乙酯和EPA的TMS酯之间提供基线分离的不同柱(30米DB-1)的仪器上。然而,由于此方法采用热注入口以在柱上将样品气化,所以它不能用来确定甘油单酯、甘油二酯或甘油三酯或者不能逃离注入口的其他重质组分的量。部分V.乙醇萃取的海藻油的表征和化学分析及其使用2,2-二甲氧基丙烷的干燥概述:此简短研究证实,AuroraInc.的乙醇萃取的海藻油的样品(20.45重量%乙醇;4.35重量%水)可以仅通过将其与2,2-二甲氧基丙烷加热并且经由蒸发除去副产物甲醇和丙酮而化学干燥至大约0.5重量%残余水。2,2-二甲氧基丙烷需要以与存在的水相等的摩尔当量使用。此工艺选择后来证实当工艺的第一个步骤从酯交换变化至总的水溶液皂化时是不必要的。仅酯交换步骤获益于干燥海藻油。皂化需要水。还获得此海藻油样品的化学分析。数据表明使用乙醇作为萃取溶剂导致大量的氯溶解到萃取的海藻油中。这最可能的是来自其中藻类生长的盐水的氯化钠(NaCl)。介绍:缩酮可以通过酸催化的水解转化回它们的母体醇和起始酮。因此,从备选角度看此反应,缩酮可以用来反应性地从宽泛的各种各样的湿有机混合物除去水。因此,我们已使用了2,2-二甲氧基丙烷以容易地干燥乙醇萃取的海藻油至大约0.4-0.6重量%残余水。此反应在缩酮和水两个方面都是化学计量的,所以对于良好结果,需要足够浓度的缩酮。此反应的副产物是甲醇和丙酮,经由蒸发易于除去的两种挥发性产物。此反应通常使用痕量的强酸催化,而明显地在乙醇萃取的海藻油中已存在的游离脂肪酸足以催化此反应。在有和没有0.75%硫酸存在的情况下进行脱水时,获得相当的结果。在“干燥后”样品中存在的少量残余水(~0.5%)可以是不可用于与2,2-二甲氧基丙烷反应的“结合”水。实验:5-加仑的乙醇萃取的海藻油是用于此研究的样品的来源。总挥发物(TV):使用OHAUSMB200水分测定仪,将一式两份的海藻油样品加热至恒定残余物重量。两次分析的平均值表明该海藻油的总挥发物含量为24.78%。这被认为是水和乙醇的组合。水含量(WC):海藻油的水含量获自溶解在无水乙醇中的一式两份海藻油样品的Karl-Fisher分析(水含量0.07%,经由Karl-Fisher分析)。使用乙醇溶剂是必要的,因为尝试将样品直接添加至Karl-Fisher设备很难。一式两份分析表明海藻油的水含量为4.35重量%。乙醇含量(EC):乙醇含量经由计算确定,即EC=TV-WC。因此,确定残余乙醇含量为20.45重量%。化学分析:将此相同海藻油的样品发送用于元素分析。要求对碳、氢、氧、氮、磷、硫、氯和残余灰分分析。分析结果在下表中给出。由于我们知晓样品含有20.45重量%乙醇和4.35重量%水,所以我们可以使用这些值来获得对于该“干燥”海藻油的校正重量百分比。这些值在“校正重量%#1”栏中给出。假定氯和灰分是无机物而不是海藻油“本身”的组分,则我们可以进一步校正所述数据以得到在“校正重量%#2”栏中给出的重量%。这应该是最具代表性的乙醇可溶性海藻油组分本身。乙醇萃取的海藻油的化学分析。元素重量%校正*重量%#1校正**重量%#2碳55.9860.7165.85氢10.199.4510.25氧24.5818.2219.76氮2.202.953.20磷0.2250.300.33硫0.4240.570.62氯2.973.98总灰分2.853.82总计99.419100.00100.01*校正至无乙醇和水以及100重量%基础。**进一步校正至无氯、无灰分和100重量%基础。基于干燥、无氯和无无机物,氮值很高,为3.20重量%。使用叶绿素的分子量/结构作为模型(Nanochloropsissalina,不具有叶绿素b或c),此数值将表明多达57%的海藻油质量为叶绿素。这太高,所以N更可能是叶绿素和蛋白质的组合。蛋白质中平均氮含量更像是16%。实施例2:利用2,2-二甲氧基丙烷的海藻油脱水未催化的反应:将100.0g的海藻油样品与30.01g的2,2-二甲氧基丙烷(bp79-810C)混合并在保持为540mmHg的旋转蒸发器上温热至500C。此温度和压力将除去丙酮和甲醇,因为它们经由化学脱水形成,而留下未反应的2,2-二甲氧基丙烷以继续其与水的反应。反应混合物的样品每一小时进行分析直至水含量变为恒定。表:未催化的结果。*2个分析的平均值:**经由计算。***压力至400mmHg。脱水在约5小时达到稳态,其中残余水大约为0.60重量%。这可以为不可用于与缩酮反应的结合水的形式。催化的反应:催化的反应以类似方式进行,除了添加0.1g的浓硫酸(0.077重量%)和在5小时之后添加另外的2,2-二甲氧基丙烷(5.0g)以使某些足够的试剂仍然存在。在此添加之后观察到一些另外的水除去,但这是轻微的。残余的水继续维持在约0.40重量%,继续表明仍然有一些看起来不可用于反应的水。表:催化的结果。*2个分析的平均值:**经由计算。***添加另外的5g2,2-二甲氧基丙烷。利用硫酸催化脱水确实增大脱水速率和可能的程度,但也许不足以对其使用的另外难题(成本,需要将其从反应混合物除去,催化自残余乙醇的二乙醚形成的可能性等)作出解释。部分VI.酯化样品的分析接收二十二个样品用于Karl-Fisher水分析和GC分析。充分振荡的样品用于Karl-Fisher分析。此时,利用在ASTMD6584(EN14105)中使用的典型TMS衍生化方法,GC分析使用柱上注入进行。为了节省分析成本,不分析所有样品。通过在50℃的水浴中温热以使样品液化,将样品制备用于GC分析。将十(10)ml的样品连同10ml的己烷添加至离心管。在剧烈振荡一分钟之后,将样品离心15分钟并将所得到的顶部己烷层取样用于衍生化GC分析。对于这些样品的结果在下表中报告。表:水浓度、酯/FFA比率和GC编号汇编2’-两小时样品。可以从这些数据得出若干结论。pH必须为或低于1.69以获得95%酯选择性。在至少95%的酯选择性的情况下所有的水浓度为5%以上。对于此高酯选择性,这是太多的水。这或许是存在的硫酸占用水,防止它进入酯化平衡的第一个迹象。甘油在低pH分解。已知的是,甘油可以脱水成为丙烯醛。在此酸性环境中,丙烯醛不大可能幸存而将有可能进一步反应,最可能是聚合。Rxn13不能已经是酯化。它看起来已经皂化。样品的分析:在单个密封塑料测试管中接收的十二个样品。样品中的十个来自在范围为从2.50下降至0.46的不同pH条件下使用乙醇在Aurora进行的酯化。一个样品是从低pH酯化中的一个回收的乙醇并且最后一个是已除去了乙醇的浓缩反应混合物。考察的目的是确定残余水含量和对于每个酯化的酯与FFA比率以及确定在回收的乙醇样品中是否存在二乙醚(DEE),并且如果存在,则存在多少二乙醚(DEE)。检查已除去乙醇的样品的酯与FFA比率将会确定是否在该乙醇蒸发期间发生了任何逆转。由于使用可以分离并定量EPA乙酯及其三甲基甲硅烷基(TMS)酯的GC方法,确定酯化反应混合物中的酯与FFA比率相对容易并且直接。通过分析这两个峰,我们可以容易地确定酯化的程度。实验结果和结论:将前11个样品通过将它们放入在50℃的水浴中达30分钟而液化。然后将它们去除,充分振荡并使用Karl-fisher分析一式两份地对水进行分析。另外,将各自5.0g样品连同10cc的己烷添加至50cc离心管,剧烈振荡然后离心5min以分离己烷和水层。此步骤从含酯/FFA的顶部己烷层分离乙醇/水/酸组分,其形成重质底相。对于每个样品去除两ml的顶部己烷层的样品并使用TMS衍生化方法经由柱上GC进行分析。对于每个样品的酯/FFA比率也报告在下表中。此时,这些比率直接从EPA乙酯峰的面积百分比和EPA本身的TMS酯的面积百分比计算。这两个峰不是分开的“基线”,所以在比率数值方面,尤其是在高酯比率下存在不确定性。非常有可能的是,报告为96.0%以上的值实际上显著比其更好。另外,在这些数值中没有对于EPA乙酯和EPA的TMS酯的相对响应因子(RFF)中的差异进行校正。而且,此时,乙基Are和EPA乙酯共洗脱,所以两种酯表示为EPA乙酯。使用分流/无分流GC方法和给出EPA的乙酯和TMS酯之间的良好、基线分离的不同柱。甚至后一种方法-柱组合也将分离ARA和EPA酯。经由柱上方法看起来为96%的比率当根据此第二种方法分析时增大至97.5%+。不幸地,这些具体样品都没有使用第二种柱/方法重新分析,所以没有可用的准确对比数值,但我确信在酯的比率方面总体增大,从96%范围至至少97.5%。表:水浓度、酯/FFA比率和GC编号汇编。样品#pHKF-1KF-2KF-平均值GC-编号酯/FFA*12.502.062.112.09207-6-148.322.092.272.372.32207-6-255.231.902.952.802.88207-6-372.941.583.083.153.12207-6-491.751.353.313.263.29207-6-596.561.123.503.403.45207-6-696.370.913.533.573.55207-6-796.680.723.893.823.86207-6-896.690.534.314.234.27207-6-996.6100.464.624.584.60207-6-1096.711-207-6-11**12-10.29207-6-12A数值来自两个酯峰的面积百分比的直接比率。**GC质量差,但通过检查对于两个C16酯峰的GC峰,在经由蒸馏的乙醇去除期间发生任何逆转还是不明显。从上表的数据可以形成若干其他观察笔记。酯化的平衡位置与高于约1.35的pH的反应介质的pH相关。低于该pH,所有的比率看起来相同。然而,上表还显示存在的水随着pH降低而增大。这似乎与酯/FFA比率随着pH降低而提高相矛盾。两个因子在这里起作用。较低的pH是添加的另外的硫酸的结果。硫酸是极强酸,其需要水分子用于适当水合。这些结合水分子具有降低的活性并且不参与酯/FFA平衡,如游离水分子那样。另外,在酯化期间存在两个液相。大部分的硫酸、水和大多数乙醇存在于较重的极性相,远离其中酯化实际发生的有机相。其他研究已显示,在有机相中有残余酸性,相当于250-350ppm的硫酸。其催化在该相中的酯化反应并将形成的水转移到重质极性相中,进一步促进酯化平衡。我们可以确信,这些反应混合物处于真实平衡。根据其他研究,不仅我们确实知晓使用足够的实验反应时间来达到平衡,而且样品在分析之前也在室温停放若干天。根据其他研究,我们知晓在这些pH下的酯化反应将达到平衡,即使仅在室温留置过夜。根据上表中的数据也是显而易见的是,水浓度随着pH降低而增大。这为什么发生?在这些系统中有三种可能的水来源。在浓硫酸中有一些水。所以一些水随着添加的另外的硫酸进入。然而,这是小贡献。水还作为酯化的产物之一形成。所以,随着更多的酯形成,也形成更多的水。这是其中酯平衡保持恒定的多达大约pH1.35的更重要的另外来源。第三个水来源是来自乙醇形成硫酸氢单乙酯的酯化。此反应在较低pH下更为重要。相对于pH的水浓度增大和酯转化率增大在图3和4中示出。明显的是,随着pH从2.50降低至约1.2,水快速地增加,其中它达到平衡然后在最低pH再次增加。这与初始地随着酯转化率增大的水浓度一致,在酯转化率达到平衡时达到平衡,然后随着硫酸氢单乙酯形成在最低pH变得更为重要而增大。作为酸强度和温度的函数的水和硫酸氢单乙酯的形成在单独研究中检查并且该研究的发现支持这些观察结果。然而,该研究显示,硫酸氢单乙酯和水甚至在室温形成,所以可能的是我们将在回收的乙醇中发现的一些额外水实际上在运送中的若干天期间的反应后已形成。此收集中的最后一个样品是从在实验室中进行的低pH酯化回收的乙醇的样品。通过Karl-fisher对水进行分析并且发现含有10.29%的水。GC分析还鉴定在乙醇以96.99%存在的情况下存在DEE,为2.13面积百分比。剩余的1%由一个较大和5个显著更小的峰组成。采用GC/MS来鉴定较大的小峰(0.45%)为乙酸乙酯。丙酸乙酯和丁酸乙酯也在小峰中被鉴定。此样品也使用TC检测器GC(207-6-12B)对水进行分析并且发现为10.16%,与通过Karl-Fisher较早发现的10.29%一致。乙酸乙酯、丙酸乙酯和丁酸乙酯的存在或许没那么令人惊讶。熟知的是,细菌存在于收获的藻类并且这些细菌可以是厌氧菌或兼性厌氧菌。这样的细菌经由厌氧发酵途径代谢糖类并且它们的产物是乙酸和丙酸为主要组分的挥发性脂肪酸(VFA)家族。它们的乙酯在酯化期间连同FFA’s的那些酯一起形成,并且由于它们具有低沸点,所以它们在蒸馏期间伴随乙醇。样品中的七个是需要衍生化GC分析的己烷萃取物;另外八个样品是从各个酯化运行回收的乙醇的样品。将它们通过KarlFisher对水进行分析并且经由GC检查它们的二乙醚含量。结果在下表中给出。样品的衍生化GC分析。样品的KarlFisher分析。8个另外的样品通过GC分析。RXN22NEUJ:此样品的柱上分析没有给出令人感兴趣的GC峰。RXN22ESTK:柱上GC分析显示一些残余重脂质仍保持(VVA+=6.91-“完全”转化率据信为约4.0%)同时在分流注入柱上的分析显示优异的酯化选择性,对于C16和EPA分别为99.1%和99.2%。RUN22EXT1M、RUN22EXT2N、RUN22EXT3O、RUN22COMP:这四个样品在一起报告,因为它们经由柱上GC分析都是几乎相同并且看起非常像RXN22ESTK的柱上GC分析。所以在萃取/水洗涤期间没有异常发生。没有明显的逆转。这对于Run22QuenQ同样是真的。它如同这其他五个GC扫描。对于所有这些样品的VVA+数值的范围为6-9面积%。此范围不是不常见的。对于在几天内重复若干次的样品,基线变化可以容易地给出此分析范围。RXN22CRFAEE:此样品通过柱上并且经由分流注入进行分析。在柱上,其看起来像已经描述的其他样品。VVA+为6.9面积%。酯化选择性(分流注入)对于C16和EPA物质分别保持为99.3%和99.4%。这些数值与起始酯化RXN22ESTK几乎不可区分。较大规模批次样品-NEUA:这是已被酸化以将皂转化成它们的游离脂肪酸形式的皂化反应混合物的样品。并且此样品中的大多数组分是FFA’s。仅约3-5%的物质保持为其乙酯形式。这是由于在原始海藻油样品中存在乙醇并且存在不足的时间或不足的水来完全水解这些残余乙酯至它们的皂形式。当然,乙酯形式经由海藻油甘油酯与乙醇的碱催化酯交换形式。这是典型的连同皂化一起发生的“生物柴油”化学。此样品在柱上分析以确定皂化是否完成并且基于GC扫描的重脂质部分的检查,转化率看起来优异,几乎完全。VVA+数值,19.5至22.5分钟之间的面积百分比的总和仅为4.18%,大约是如我们曾经见过的VVA+值一样低。结论-在工艺中的成功第一步骤。较大规模批次样品-EXT1D、Ext2E、Ext3F和EXT4G:这些是中和的皂化反应混合物的连续庚烷萃取样品。它们具有与之前样品相同的GC谱图,因为它们应该具有。这里重要的是确定需要多少次萃取来回收存在的所有FFA’s。当然,萃取将非极性产物与皂化/中和的极性组分(乙醇、水、甘油、残余酸、K2SO4盐和可能存在的任何其他极性物质)分开。因为所有4个样品以相同方式处理和制备用于分析,所以我们可以通过根据连续萃取的色谱图的总面积量测量萃取效率。对于4个连续萃取,它们分别为7.25、2.28、0.89和0.32(全部xe7)。对于四个萃取,%回收率为68%、21%、8.4%和2.1%。最后一个GC几乎没有显示任何FFA峰。三个萃取看起来足以用于FFA回收。较大规模批次样品ESTB:使用柱上的GC分析对于此样品总体上是不令人满意的。C16峰计算为98.2%酯选择性而EPA峰仅给出86.2%。这是不合理的。为了确定由C16峰给出的高酯化选择性,使用分流注入方法对样品重新分析。对于C16和EPA物质,酯化选择性现在计算为99.2%和99.3%。这代表优异的酯化性能。较大规模批次样品-MIXESTH:样品非常密切地类似于之前ESTB样品的柱上GC扫描。较大规模批次样品-STRIPI:此样品是来自一些庚烷溶剂已经经由蒸发除去的酯化或水洗酯化的样品。较大规模批次样品-CRFAEE:这是最可能的最终汽提乙酯产物的样品。将其使用两种GC方法进行分析。使用分流注入分析,乙酯化选择性利用C16或EPA峰计算为99.0%。将这些值往回与ESTB的那些值(对于C16和EPA分别为99.2%和99.3%)进行比较,看起来在酯化在单元式操作期间很少或没有逆转发生。这是好消息并且在最终粗产物中的最终99%酯化选择性是优异的。基于这些样品的分析,随后的大规模运行应产生相当质量的粗乙酯,提供如同实验室试验的大规模运行。将在中试工厂中制备的粗海藻油乙酯发送至分馏。AUR_样品1:此样品包括粗海藻油乙酯。利用C16和EPAGC峰,酯选择性计算分别为97.3%和97.0%。存在的胆固醇为3.8面积%。从用作内标的月桂酸甲酯,看起来此样品为约5.7重量%EPA乙酯。它可以是脱挥发样品,预备用于分馏。AUR_样品2:GC扫描看起来与之前的样品的GC扫描相同。在以上讨论的相同C16和EPA基础上,酯选择性少量地增大至97.5%和97.9%。此样品可以更深层地脱挥发。AUR_样品3:此样品是几乎除去全部乙酯的“尾分”样品,假定经由在高温度和低压力下挥发。样品在胆固醇方面高(16.4面积%),存在许多其他重脂质(VVA+=31.8面积%)。一些残余乙酯在C16下保持为约1面积%并且在EPA乙酯下为约3面积%。如果需要,此样品可以重新皂化、中和并且萃取以确定其含有多少可用的脂质。AUR_样品4:此材料是蒸发并重新浓缩的材料,其之前的样品是非挥发的尾分。它几乎没有胆固醇(<1.0面积%)并且不具有重脂质。对于C16和EPA物质,酯选择性分别保持为97.5%和97.9%,所以这种分离没有破坏任何乙酯或FFA。这些是几乎是与对于AUR_样品2获得的相同值。实施例3:行动4-在2通道中利用乙醇的连续转化A.概述转化是下游藻类工艺中的第二步骤。其消耗粗海藻油并将其转化为粗脂肪酸乙酯,其可以进一步精制以生产65%EPA-EE以及其他产物。对于行动4,将工艺重新设计为使用大部分来自行动3的相同设备的两通道系统。用于此工艺的进料是在乙醇中的粗海藻油萃取物。在这种情况下该油仅具有20%乙醇而不是如行动3中的50%。一号通道包括在水中的碱(氢氧化钾)催化的皂化接着用硫酸中和。然后将该材料庚烷萃取异在进入步骤2之前纯化脂质流。在萃取后还有倾析步骤以帮助除去在塔顶馏出物中发现且在工艺中后面引起潜在问题的低密度固体。通道2使用来自通道1的最终材料作为进料。其主要由溶解在庚烷中的游离脂肪酸(FFA)组成。第一步骤是使用乙醇和硫酸将它们酯化为脂肪酸乙酯(FAEE)。此材料然后水洗以除去酸和乙醇。倾析器在此水洗后以放出任何废弃沉淀。除去庚烷,留下最终转化产物,我们称其为粗FAEE。在该运行期间,加工加热箱添加至用于庚烷萃取和水洗的karr柱以有助于相分离。运行行动4以证实来自行动3的工艺改变将能够实现稳态连续转化运行。还进行其以使物料进一步加工以最终用各种各样的验证试验,包括FDA的通常视为安全试验(GRAS)。进料材料具有20%乙醇而不是50%,这是由于需要水代替乙醇的工艺中的化学改变所致。之前水受限,因为它将生成FFA;然而,发现限制水仅在获得高转化率方面有点成功。改变该工艺以首先有目的地经由在水中的皂化制备所有FFA,因为这是一个容易的反应以推动至完成,然后经由庚烷萃取除去水并经由在乙醇和硫酸中的无水酯化将所有FFA转化为FAEE。萃取温热运行以增加萃取效率和相分离。在萃取之后,将乙醇和酸用在循环汽提系统中的水和庚烷洗出以生产最终转化产物从而发送以用于精制。从工艺前方开始,粗品需要在其在的按重量计至少10%乙醇以能够实现混合并因此实现皂化中的反应。中和需要获得约1.5的pH以能够实现庚烷有效地萃取FFA。萃取必须温热>60℃进行以获得良好萃取效率并且萃取的塔顶馏出物需要倾析以除去形成的低密度水污泥。此污泥不应装入酯化反应器中,因为它可以引起堵塞并且在一些情况下低转化率。发现较差转化的材料可以通过除去水层并将其放回到进料桶中进行再加工。除了在最终停车期间,酯化后水洗顺利地运行。固体在界面处堆积,当材料排出时引起堵塞。向前,分批水洗将更好工作以防止此堆积。庚烷汽提如计划进行。第二汽提环将有助于减少产物中保留的挥发物。在工艺的大部分中,保持物料流动和温热是重要的或者在管线堵塞和淤塞(fouling)的情况下将有问题。当在多数步骤2期间油是在庚烷中稀释的时这不太是问题。为防止这点,任何时间装置停机非常长时间时,管线应该用溶剂或水冲洗。流动蒸气清洁由这些液体留下的东西中的大多数。B.介绍在行动3中,我们确定在实验室开发的乙醇工艺下的一些问题并在常规工艺上解决以运行行动4。来自行动3的设备能够再利用用于此运行。运行此行动以尝试和证实该工艺可以运行。将其设计为在一些化学变化和一些轻微管道工程变化的情况下对每个通道使用几乎全部相同的设备的2通道系统。来自行动3的大变化是含水第一通道。发现使用直接来自粗海藻油的乙醇和酸的酯化是难以推动完成的慢工艺。然而,在所述脂质为FFA形式之后,其更易于酯化。幸运地,直接至FFA的反应使用水和强碱是简单且快速完成。萃取将正好如同利用EE一样利用FFA进行,并且它除去进入酯化的水,其在那里应增大转化率。在实验室,确定了相比于干燥粗油,皂化需要按质量计18%KOH。进行中和几乎下降至约1.5的pH。然而,对于在该行动中使用的确定设置为至少低于3。利用庚烷测试萃取,但设置没有广泛地测试,因为逆流萃取柱很难在实验室准确地建模。因此使用中试工厂来确定这些设置。广泛地测试酯化。确定了相比于在庚烷流中的15%油,酯化需要按质量计3%酸。之前发现需要2:1乙醇对酸的溶液来最小化稀释中的水形成。此两步工艺显示在图5和6中。此工艺的第一步是皂化(图5)。将50%KOH水溶液与海藻油组合以生成皂。这作为栓塞流反应在管线中进行。蒸汽加热的同心管换热器提供用于反应的所需温度。在换热器之后,有一个小罐以增加反应的停留时间。下表显示用于此部分的反应的操作条件。在下面的许多操作条件表中值得注意的一件事情是进料速率具有泵设置和实际数值二者。这是由于不准确的泵校准。如果在将来使用相同的泵,则泵设置在那里作为设置点的提醒,否则实际的kg/hr是应该作为目标的值。在运行期间确定pH>14。这实际上高于大多数pH探针的读出极限。在我们的系统中,基于具有20%乙醇的油进料的18%KOH结束为>14。此操作条件只是一种检查以确信流动是平衡的。进料中的挥发物也是在运行期间确定的事情,但是那将在报告的后面讨论。表:皂化稳态操作条件。在皂化之后,将皂进行皂化以将它们转化为非极性FFA’s。这有助于后面的萃取,因为与皂不同,相比于水,FFA更可溶于庚烷。将酸在稀释反应器中与水预混合,然后添加至海藻油、KOH混合物。目标是在水溶液中的10%酸。由于酸泵上的下限问题,其更接近12.5%运行。酸和水溢出到中和反应器中,并且皂化的海藻油从皂化流入。中和与稀释反应器具有连续蒸汽空间,其将它们保持在相同压力。在顶部上有冷凝器,其将来自任一反应器的任何蒸汽冷凝回到稀释反应器中。对两个反应器搅拌以确保适当混合。也不应该运行足够高以形成涡流,因为那可以导致水平读数误差。涡流将是显而易见的,因为水平读出将变得不稳定,并且反应器经常将开始振荡。稀释反应器上的冷却水将反应器保持在约室温以下。使用的冷却水保持在5℃。中和反应器上的蒸汽套控制器温度。下表显示在行动4期间运行的稳态操作条件。表:用于中和的稳态操作条件。中和目标pH1.5温度75℃压力3barg来自皂化的进料9.07kg/hr酸进料:泵设置0.75L/hr实际的1.05kg/hr水进料:泵设置5.00L/hr实际的7.00kg/hr目标目标40%停留时间60分钟中和的混合物保持在中间槽中,然后泵入到Karr柱中用于庚烷萃取。中间槽允许吸收小工艺失误(upset)而没有完全停车。中间槽具有外部循环回路以防止固体沉淀并堵塞槽出口。为了在酯化之前除去水以及其他不期望物料如盐、甘油和来自海藻油流的固体,将中和的海藻油混合物进料到称为Karr柱的搅拌逆流液体-液体萃取柱中。图5显示进入底部的庚烷、在顶部的水和进入中部的中和海藻油。使用的Karr柱直径为2,具有12ft的搅拌和在每一端上的4L分离圆顶(disengagingbell)。海藻油混合物实际上从顶部分离圆顶下方约2英尺进料。水经管道向上以运行到顶部中;然而它不用于大多数的运行,所以在下表中发现的稳态操作条件不包括水进料。通过将绝缘纸板置于柱周围的箱中并从上方单元式加热器形成通风管线至该箱。监测温度并通过操控隔板以将热空气重新引导至或远离Karr柱而控制。这足够良好工作以将系统控制在几度内。然而,在将来,应该可能采用具有更少操作者集约控制的系统。为了容易地启动Karr柱,首先进料庚烷以填充该柱,然后添加油混合物。表:用于Karr庚烷萃取柱的稳态操作条件。萃取温度60-70℃来自中间槽的进料:泵设置4L/hr实际的6.01kg/hr庚烷进料:泵设置17L/hr实际的6.38kg/hr庚烷中的目标油含量15%质量界面控制350%塔顶馏出物排放7.34kg/hr尾分排放5.05kg/hr搅拌200次/minKarr柱的塔顶馏出物通过管道进入倾析器以允许更多沉降时间来去除任何固体、水或来到塔顶馏出物的其他废弃物。来自倾析器的塔顶馏出物然后泵入一定规模的桶中,当满时,将其泵入到桶中以用作用于步骤#2的进料。karr和倾析器二者的尾分泵入废物桶中。Karr尾分出口速率使用泵初始地控制。在步骤2(图6)中,当尾分含有很少固体时,在其位置使用自动阀,因为所述泵不会保持压力。记录对于从系统除去的所有桶或添加至系统的海藻油的重量用于质量平衡目的。对于两个步骤,在启动期间每一小时或每两小时或在稳态后每3小时或每6小时对最大混合流运行分析。这是为了确保获得适当的条件。来自步骤1的产物:溶解在庚烷中的FFA储存在桶总同时重置系统用于步骤2。将系统排空并用水、乙醇、庚烷或蒸气冲洗以除去大部分的固体和积油。Karr柱上的管道设施切换至使得油将在底部进料而不是从距离顶部下方一点进料。而且,庚烷汽提系统安装管道,在倾析器塔顶馏出物收集桶出具有进料点。图6显示用于步骤2的流程图。其非常类似于步骤1,在流标签方面具有少量变化和轻微管道设施差异。来自步骤1的产物,在庚烷中的FFA,使用如之前的相同进料系统进料到步骤2的起点。使用皂化反应器的换热器来预热庚烷以使其进入已经热的酯化反应器。酸好乙醇在酸稀释反应器中进行混合,该酸稀释反应器保持在或低于25℃以防止水形成。然后将此混合物也进料到酯化中以开始反应。此反应在压力下完成以能够实现95℃的较高反应温度。该反应可以在较低温度下实现,但是花更多的时间。所以,高温用来减少停留时间并且增大生产能力。用于酯化的稳态操作条件可以在下表中找到。当将进料物料泵送到系统中时确保在桶的底部上没有沉积污泥层是很重要的。如果有污泥,则在反应之前进料需要倾析。污泥可以降低反应转化率以及在小管线中导致堵塞问题。表:用于酯化的稳态操作条件。将酯化混合物进料到作为储存点的中间槽中,然后将其进料到Karr柱中用于水洗步骤。进行水洗以除去酸和乙醇,然后庚烷可以汽提。这防止后来在处理中逆转。在将油、庚烷、酸、乙醇混合物进料到底部并且水进料到顶部的搅拌Karr柱中逆流地进行水洗。将乙醇、水和酸从底部去除并且在庚烷中的转化油从顶部溢出。底部的进料速率通过dP水平控制器控制,该控制器测量底部圆顶中的界面水平。其设置有泵和分离管线,其中在其上的阀作为备份。底部出来的物料具有使使用中的泵结构的趋势。在泵停止工作后该阀很好工作并且用于剩余的运行。用于水洗的设置可以在下表中找到。在最后庚烷汽提之前,物料经过倾析步骤。如同步骤1中一样,工艺首先利用庚烷相进料开始。在这种情况下是油流。在水开始允许物料的一些洗涤之前,其填充约一半,然后其溢流到倾析器中。表:对于水洗的稳态操作条件。水洗塔顶馏出物的目标pH>3来自酯化的进料:泵设置6L/hr实际的15.32kg/hr水进料:泵设置2L/hr实际的1.09kg/hr温度65℃界面控制400%塔顶馏出物排放13.81kg/hr尾分排放2.60kg/hr搅拌200次/min转化工艺中的最后单元式操作是庚烷汽提。这进行以在进入精制工艺之前得到尽可能低的庚烷。它们运行最后脱溶剂化,但是可以仅处理少量溶剂。而且,溶解在大量溶剂中的运输材料是显著更有害的并且显著更昂贵的。庚烷汽提系统发送通过回路周围的泵收集的Karr塔顶馏出物。此回路包含换热器随后的分离罐,在那里庚烷当其随着合适温度并且系统在适量真空下时闪蒸出。然后物料循环通过系统。在系统处于稳态后,少量总是泵出底部。同样地,新的物料总是进料。这使得能够连续庚烷汽提。系统构建在第三层上,其中循环泵在第一层上。这为泵提供足够的排出压力,从而即使在低压操作条件下也保持其泵送。通过进料和汽提将其启动直至建立足够的水平,并且物料处于合适挥发物含量以开始出料。通过将物料冷凝到罐中来收集庚烷。下方有第二个罐,其如同压力分离罐一样工作,使得不需要破坏真空而将冷凝器吸入到桶中。下表显示对于庚烷汽提运行的稳态操作条件。将来自此步骤的产物流收集在20L聚丙烯桶中。在桶完成后,添加100ppm的柠檬酸和生育酚二者,然后将桶用氮气覆盖并密封。表:用于庚烷汽提的稳态操作条件。在开始行动之前,进行一些计算以确定应该花多长时间来处理所有的进料物料,在每个步骤中将花费多少时间,以及如果始终看到100%转化率和效率则值得多少物料。下表详述了此信息。表:预期的运行时间和生产。16操作天数42%步骤#1中的时间的58%步骤2中的时间的%161步骤#1中的小时223步骤#2中的小时45280%海藻油进料的Kg362海藻油进料的Kg43.4EPA进料的Kg66.865%EPA进料的Kg在沿工艺的许多不同阶段取样。样品口位于在系统的所有进口和出口上,以及设备的主要部分之间。每1-2小时对过程中的每个点取样,这取决于该单元式操作启动期间的停留时间。在一些情况下,每3然后每6取样并且将过程用线画出。对于每个流的示例性分析数据在步骤1中,不对KarlFisher或挥发物运行皂化样品,因为这些对于工厂操作不重要。对于皂化要注意的另一件事情是转化率或摩尔EE/(EE+FFA)极低。这是因为大多数脂质为FFA形式。事实上,来自该表的EE/(FFA+EE)数值越低,总体皂化转化率越好。仅在较低pH下,中和非常相似。色谱图实际上几乎相同,因为必须进行中和以在GC上运行皂化样品。karr塔顶馏出物仅是中和混合物的庚烷萃取。低的水显示发生良好的清洁萃取而没有太多的废弃物来到塔顶馏出物。85%至90%之间的VOC显示萃取有效地发生。目标是达到85%VOC,但那并不总是实现。karr尾分上的VOC实际上是利用庚烷的同等重量反萃取的VOC,以确保没有残留脂质,所得约99%的高VOC是期望的,并且大多数情况下获得。当这个数值降低时,并且塔顶馏出物挥发物升高时,其意味着萃取效率是个问题。色谱图也显示很少至没有脂质,如在%EPA栏中显示的。酯化,步骤2的第一部分通过主要关注色谱图进行控制。EE和FFA之间的比率是最重要的。目标是尽可能高的数值。通常其在95%至98%之间。此转化也通过水洗和汽提跟进以确保没有逆转。一些样品如水洗塔顶馏出物实例显示比所需的低一点,但是总体达到转化。经由KarlFisher的水含量在酯化中是重要的以确保平衡被推向乙酯。对于KF的目标是低于1%。存在失误,其中转化显著降低并且发现水增加。这就爱过你在后面进一步讨论。为了确保所有的酸、乙醇和水被冲去,在水洗塔顶馏出物上运行KarlFisher和pH。只要pH高于3并且水低于<0.1%左右,就确定洗涤是充分的。水洗尾分以与步骤1中的karr尾分的相同方式进行分析,以确保脂质不会是使其进入废物流中。表:产物:粗FAEE总的说来,从生产角度以及研究运行两方面看,此行动是成功的。以97%转化率制得69kg的粗FAEE。该物料含有17%EPA和17%庚烷。关于在上表中每一批次的称为粗FAEE的转化产物的细节在以下。测量样品中的乙酯或甲酯含量如下进行。对于在文献中发现的相对响应因子,调整来自色谱图的面积百分比以得到准确的质量%值。图7显示在无溶剂基础上的各个乙酯脂质基团的质量%。除了生产物料,此行动也是成功的,因为它比之前更顺利地进行并且对于所有单元操作实现了稳态连续操作。事实上,每个单元操作在稳态连续地运行至少24小时。步骤2比步骤1更顺利地运行。在所有步骤2系统的部件连接的情况下并且在流动平衡的情况下,记录了超过48小时的连续稳态操作。不幸地,步骤1连续稳态由于过程失误所致没有同时对反应和萃取实现。由于所述失误,步骤一花费较长时间运行并且在三周期间完成。步骤2在一周内都完成,并且从不需要全部系统停机。进行大多数停机以使单元操作之间的流动平衡。此工艺的瓶颈是汽提系统,所以有时所述反应和水洗必须停止以使汽提器跟上。完成稳态平衡和转化率计算以判断在长期连续转化运行期间所预期的结果是什么。这些计算的结果详述在下表中。每个数值计算自在此行动期间获得的在稳态运行时间段期间的许多数据点上的平均值。这去除了与启动、停机和过程失误相关的误差。因此这些数值假定工厂顺利地运行。回收率是进料和含有产物的出口流EPA含量之间的质量平衡。对于每个步骤和对于总体回收率,每个步骤的回收率相乘在一起。损失率是相比于单位操作进料流,来自废物出口流中的多少EPA的比较。转化率关注在工艺的该点处多少摩尔百分比的EPA处于所需形式。对于皂化,其是FFA,并且在酯化之后,其是EE。转化率对于不同于实际反应的操作很重要以确保没有逆转发生。为了计算对于通道2的%转化率,对单个步骤转化率求平均值。然后对于总的转化率,将其与来自通道#1的转化率相乘。由于通道2中的每个步骤不是反应,所以那里的转化率只是确保没有逆反应。这就是为什么对来自通道2中的每个步骤的转化率求平均值而不是相乘。表:对于EPA的稳态回收率、损失率和转化率计算。通过取总体回收率并将其乘以%转化率,可以确定在类似于在行动4中运行的工厂构建的连续转化工厂中,可以在稳态运行的同时获得95.8%的收率。EPA和其他脂质含量信息及其处于什么形式来自在工厂运行期间运行的色谱图。对于转化过程最有用的色谱图是在运行气相色谱法之前通过将FFA转化为TMS酯产生的。然后这些色谱图显示FFA’s、EE’s和留下未转化的任何重脂质,以及值得注意的一些其他峰。单个组分的定量通过在分析之前向样品中添加内标月桂酸甲酯和已知的浓度进行。皂化色谱图比酯化色谱图稍微更难于定量地解释。这是由于这样的事实所致,即在反应之前,脂质为多种多样的形式,而不是仅为如同反应之后的一种形式。这意味着,为了准确地定量进料中的EPA,要么必须鉴别所有的形式并定量,然后添加在一起,要么所述物料必须进行反应然后定量。这也意味着未转化的脂质难以在皂化样品上进行定量。所以,开发了创新方法来评估反应的程度。转化率的测量以成对方式进行,所述方式都用来获得转化率数值。皂化样品必须首先进行中和以制备FFA,然后可以进行分析。因此,皂化样品可以要么是来自工厂的中和样品要么是在实验室中中和的实际皂化样品。对于必须添加用于中和目的的任何另外地物料,对样品质量%进行校正。对于皂化关注的第一件事是在色谱图的最右侧上的重脂质面积。在那里有许多不同的峰,其使得难以准确地定量。从约19至23分钟的该面积的大部分的积分得到我们称为我们的“重脂质峰”的面积,此面积随着反应进行和重脂质变为FFA而将减小。发现在“重脂质峰”面积数大约与在约18分钟在其旁边出现的胆固醇相同后,反应完成。在此色谱图上要关注的另一件事是EPAFFA峰相对EPAEE峰。由于随着近来有乙醇进入,在皂化步骤期间将生成进料EE。然而,只要仍然有足够的强碱和水存在,则他们将转化为FFA。图9提供在皂化期间发生的反应的示意图。这意味着如果有EE留下,则就没有足够的主要反应物之一来将一切推向FFA,所以最可能的是也有未转化的重脂质。如果不是由于EE的存在潜在地预示的未转化的重脂质,则在此时具有一些EE将是美好的,因为那是最终目标。到皂的反应与到乙酯的反应竞争,意味着它们都是同时发生。然而,在脂质变为乙酯后,如果仍然有水和碱,则它也将转化为皂。所以色谱图中缺少乙酯显示有足够的碱和水来完成两个反应路径从而仅获得皂。对于大多数的过程样品,使用重脂质峰和乙酯峰来确定皂化是否如所期望的那样发生。然而,为了确保我们能够显示完全转化,将这些样品中的一些进一步分析。取来自中试工厂的一些良好皂化的样品并进一步反应。这通过添加更多的碱并加热延长的时间段进行。想法是使样品完全反应然后将两个色谱图进行对比。来自这些样品之一的色谱图在图10中下方重叠。由在此色谱图中的两个峰几乎完全重叠,特别是在重脂质区域中,所以它显示在中试工厂中实现完全转化。所述色谱图显示重脂质峰和乙酯峰也完全转化。即使皂化转化率不是最大定量的数值,我们也可以证实最小转化率数值。因为仅EE或FFA形式的EPA是可鉴别的并且因此在色谱图上是可定量的,所以转化率必须是至少与摩尔回收率一样高。因此对于皂化,摩尔回收率为98.86%。这意味着我们要么在98.86%回收率获得100%转化率,在100%质量回收率的98.86%转化率,要么是其间的相乘得到98.86%的一个数值。对于酯化的转化率计算稍微更直接。这是由于这样的事实所致,即与在皂化中不同,脂质在反应前后都处于易于可测量形式。所述酯化将在皂化/中和中形成的FFA转化为乙酯。对于转化率的计算通过找到处于乙酯形式的EPA的摩尔百分比进行。这可以通过等式#1描述。酯化反应色谱图的视图示于图11。等式#1:转化率=摩尔%EPA-EE/(摩尔%EPA-EE+摩尔%EPA-FFA)为了从色谱图得到每种形式的摩尔%EPA,需要一些工作。对此的计算可以通过等式#2描述。为了完成,利用对于FFA的数据,EPA-FFA代替所有的EPA-EE。这对于C16或任何其他脂质同样适用。转化以摩尔%完成,因为不同脂质形式具有不同的分子量,所以技术上按质量%从FFA到EE可以以几乎105%转化率完成。等式#2:摩尔%EPA-EE=(样品中IS浓度x面积数EPA-EEx相对响应因子)/(IS面积数x样品质量x稀释因子x脂质的MW)稀释因子是在添加内标之前多少样品通过溶剂或其他添加剂稀释的计算。内标缩写为IS。相对响应因子是计算数值,其显示相比于在相同浓度下的内标响应,对于每种脂质在色谱图上将有多少更大或更小的信号。这些相对响应因子通过如下方式确定,即在柱上以已知浓度运行一系列标准(一个用于关注的每种脂质)并判断它们的面积数如何与将使用的内标相关。每种脂质的质量%对于能够在系统上运行质量平衡并确保脂质不损失也是重要的。等式2可以通过从分母去除脂质的MW而变为任何脂质的质量%。这些重叠的色谱图显示显著更容易解释变化。蓝色信号是酯化前样品,而红色是酯化后的。乙酯峰显示在色谱图中比它们的TMS酯对应部分稍微较早。记住TMS酯是FFA已被官能化以用于可视化。红色信号看起来好像它已向左边稍微移动,除了在左边的第一个峰、内标峰。这显示FFA转化为乙酯。红色信号中缺少FFA峰显示转化完全。通过在程序工作站中将这些峰下方的面积求积分,并使用等式#1和#2,可以对反应的物料计算摩尔转化率。并且可以计算每个样品的质量%用于收率/回收率计算。对于稳态操作的所有这些计算的最终平均值在以上显示。对于来自萃取、水洗和庚烷汽提的样品也运行色谱法。将这些连同流动数据一起用来计算质量回收率。这些色谱图的实例可以在附录中找到,尽管它们看起来与它们之前的反应样品类似。损失率计算基于用于萃取和水洗步骤的废物流的色谱图。所述色谱图在图12的下方。这些色谱图都不显示任何脂质,这意味着在任一这些样品中都没有脂质损失。即使在此操作的稳态运行期间,也存在具有极小脂质峰的少数Karr尾分样品,其导致轻微的总体损失。然而,其仍然是极小的,总计总体损失低于1%,见表。步骤1发现在行动期间,存在许多小的失误和一些重大的失误,其使得我们发现之前不知晓的重要操作条件。以粗油开始,发现将冷的粗品铲入进料桶中,相比于如果将其加热并泵入,从该桶能够实现更大的回收率。这可能是在此行动中,由于粗油包装在顶部打开桶中而不是具有桶孔的桶中。在粗品在进料桶中之后,将其加热。然而这确实意味着在进料至皂化方面有突破,因为试图在所有粗品是液体之前启动泵,导致一些管线堵塞问题,并且在一个点处导致泵故障。此堵塞问题对于槽循环泵和油进料泵至皂化都一样。将粗品加热至60-70℃。这在没有使乙醇太快地风化的情况下将其保持为液体并且可泵送。在这些温度下一周静置结束时,EPA%没有降低,这意味着其在该时间段是稳定的。除了在第一次启动中的小问题和被快速修复(fix)的一个中间过程失误,皂化顺利地运行。在第一次启动时,首先启动油,然后是KOH。整个管线被非常实心的物料堵塞。管线必须破坏并用金属棒推出所述物料。据推断粗品已足够冷却以启动栓塞然后与KOH停留在管线中导致混合物过度反应并固化。之后,步骤启动KOH然后插入油中。这对于其余的运行良好工作。大的失误发生在步骤1的最后一周中。皂化样品开始看起来非常稠。混合物的色谱图显示极低的转化率。当反应良好进行时,样品为更多液体,并且可倾倒。已发现粗品进料中的挥发物从原始20%下降至8%。添加更多的乙醇并且这似乎修复该问题,使挥发物回到高于10%。理论是乙醇充当油和KOH水溶液之间的共溶剂。如果粗品和KOH以及水没有适当混合,则反应不能发生。乙醇充当混合剂。对此的另一种可能修复是将一些皂化的物料循环回到进料中。所述皂将充当共溶剂。这是在工厂的原始设计中,但在许多迭代和变化之后,用于循环的目的被遗忘并去除,其是增加的复杂性。在乙醇含量回到高于10%之后,反应如预期地进行。这是表1中10-20%乙醇的皂化进料规格源自的地方。20%乙醇的极限是由于以下事实所致,即额外的乙醇将仅使得后来更难的、能量密集型的乙醇回收。当皂化反应由于进料中的低乙醇所致没有进行时,注意到在萃取步骤中在尾分和塔顶馏出物都有更多的污泥。确定了这可能是未转化的油。还观察到相分离没有如在该材料经过时那么清洁,表明未转化的油可能潜在地导致乳液。这是在将来要注意的事情。中和步骤几乎是直接的。然而,它确实不太需要预期的化学计量量的硫酸来中和皂。这表明一些强碱在皂化步骤中发生反应而不是仅充当催化剂。存在与海藻油的许多潜在副反应,尤其是由于不是所有组分都被鉴别,所以这并不令人惊讶。得到的另一个发现是使用10%硫酸水溶液有意义。初始地进行稀释为在水中的20%硫酸。然而,存在固体堵塞的问题,其被确定为由于在混合物中没有溶解的在中和中形成的硫酸钾所致。增加水以补偿,使得形成的所有的盐在65℃保持为可溶。目标是10%溶液,但是实际上使用的液流更接近12.5%酸。这仍然足以溶解所有或足够的盐从而除去堵塞问题。这也结束了去除对于萃取步骤中的额外水的需要。在中和时出现的水足以洗去所有的乙醇和盐。这使得Karr柱显著更简单,仅具有两个进料点而不是3个。利用庚烷萃取得到一对重大发现。当装置首次启动时,萃取效率极低。启动搅拌有帮助,但没有让我们得到所需的水平,并且存在相分离和污泥物料来到塔顶馏出物的问题。确定热可能有助于萃取和相分离。在实验室将良好混合的样品加热,并且在样品达到55℃的时间前后,它开始迅速分离并且顶部庚烷相具有所需的油含量。基于此发现,构建“热箱”以利用单元式加热器之一对Karr柱加热。正是由绝热泡沫制成的箱经由铝空气管道附着至加热器。一系列通气孔有助于控制通过热电偶监测的温度。这修复了所述问题,并且萃取效率增大至预期的值。“热箱”在60-70℃之间运行,因为发现在连续装置中得到最佳萃取同时没有混合物沸腾的风险。图13显示加热Karr柱对于萃取效率带来的差异。蓝色是Karr塔顶馏出物的挥发物,对此的目标是85%,意味着在混合物中有15%油。红色是尾分的反萃取的挥发物。这应尽可能接近100%,意味着在尾分中没有剩余脂质。数据中的间隙是当形成并安装热箱时的情形。在达到温度后,花费几小时完全管线输出,但是塔顶馏出物的VOC下降至85-90%并且塔顶馏出物都为约99%,意味着所有的脂质被萃取到庚烷中。朝着运行步骤1结束得到另一个发现。当发现皂化进料中的乙醇的问题时,其以导致所有物料分批进入到在高pH的储存槽中的方式修复。然后混合物通过使酸性水通过系统进入该相同槽并周期性地取样直至pH低于3进行中和。这是对于任何以FFA形式而不是作为皂所需的理论pH,然而该pH在大多数实验中运行并且在行动早期更低。这里获得的pH为2.7。当就爱那个物料进料至karr柱时,萃取效率再次下降。为了补偿,添加更多的酸以将pH进一步降低至2.14。这改善了萃取,但是其仍然低。结束工作的pH为1.6。这是对于表2中的中和的pH操作条件的来源。它可以在较高pH下工作,但是需要更多的实验。图14显示通过挥发物含量相对于进料的pH测量的萃取效率的趋势。容易看到,随着进料的pH降低,Karr塔顶馏出物中的挥发物含量下降,并且尾分的挥发物增加。这两种趋势的组合显示萃取效率随着pH下降而增大,并且为了确保良好的效率,pH必须为约1.5。应当再次注意,尾分挥发物真实地是尾分反萃取的挥发物。这在某些方面是困难的试验,因为如果萃取效率差,尤其是因为pH,则就没有实验室中萃取将更好一些工作的理由,所以可能在尾分中有比此VOC更多的脂质,或者甚至GC实际地显示。然而,只要萃取良好进行,尾分的VOC检查将显示所有的脂质被去除。检查的另一种方式是通过质量平衡,其显示在稳态,此步骤中的回收率极高。因此,在尾分中很少或没有脂质损失。步骤2发现步骤2总体比步骤一更顺利地运行。在系统启动之后,从未有全部系统停机,并且真实地仅有一次失误。酯化必须连同水洗一起停机以便不会克服汽提器的容量。在再次启动后,调整流动以更好地平衡系统。在将步骤1的新桶添加至进料桶时发生失误。添加的物料也不倾析并且在底部有大量的污泥。来自酯化的色谱图开始恢复,显示越来越少的转化。停止酯化并将全部泵入中间槽中。允许该槽沉降并将污泥物料连同沉淀的酸和乙醇一起排出至废弃物。这通过计算已经添加的酸和乙醇的量并聚拢完成,其大多数除去,然后取样以肉眼地检查残余污泥。进料桶的底部也泵出以除去在其中沉淀的污泥。在这完成后,将中间槽中的物料泵回到进料槽中用于处理。之后的转化率回升至预期的水平。由此得到两个领会。一个是将污泥带入酯化对反应有害。这最可能是由于污泥中的水所致,其导致FFA和EE之间的反应平衡移动。第二个领会是物料可以再处理。这对于工艺的可靠性非常重要。如果有工艺失误,则可以将物料送至常用槽然后在再处理之前进行处理。这将意味着工艺失误将不必需地是废弃大量物料。这对于可能的间歇工艺尤其适用。如果物料不能被再处理,则将有可能破坏整个批次。在各种各样的酯化样品上运行水分析以确证关于水和低转化率的理论。初始地在低水的情况下所有数据回升。认识到由于其是两相反应,所以水将在底相中并且除非在分析之前振荡样品,则它不会显现。图15显示混合酯化样品中的水相对于转化率的曲线图。在水和低转化率之间存在明确的关联。有一些极端值但是总体地,混合酯化样品中的水必须低于5%以得到高转化率。在工艺失误期间选取具有较高水的样品,其中转化率低,并且在该事实之后运行KarlFishers以检查我们的理论。水洗难以置信地在整个时间顺利地运行。其能够启动和停机而无需任何努力。然而,当工厂停机时,在将柱从底部排出口取出时有麻烦。固定堆积在界面处,其在排出时堵塞底部端口。柱必须从侧口去除,然后固体必须破碎,然后才能够将它们除去。这使得连续水洗缺乏吸引力。已经提出此步骤间歇地进行,因为相分离如此清洁。为了有助于此,可以倾析乙醇和酸层,因为酯化是两相反应。这将降低所需的水以及使得相分流甚至更清晰。庚烷汽提也非常顺利地运行。将装置的顶部置于第三层上并将循环泵置于底层上修复在行动3中发现的真空将液体拉动远离泵并停止循环的问题。在没有循环问题的情况下获得-0.95巴的压力。这种情况下的极限是真空泵和支持系统。所述温度理论上可以升高以增大汽提速度,然而循环泵仅额定用于80℃,所以那是用于液体流的最大温度。图16提供了在运行的最后两天内在庚烷汽提器上的换热器出口温度的曲线图。庚烷汽提器的操作稳定。温度有一次微降,这可能是由于损失一个或多个沸腾器,减少到达换热器的蒸气所致。随着时间,在此曲线上,汽提器出口上的挥发物或产物都在15%至23%之间。在这个时间结束时除去的产品桶具有17.2%的平均挥发物。这一次,温度通过由控制系统确定的蒸气输入自动地控制。其他发现还得到与具体单元操作不是良好相关的其他发现。这些现在将进行讨论。全面启动和停机在极小难度下完成。像往常一样,在启动期间有泵问题。大多数那些问题是由于泵上缺少启动所致。当停机时,持续周末或由于过程失误所致,发现泵送油混合物的所有的泵以及不易于可加热的任何管线需要冲洗。我们使用和制备的材料具有在管线中堆积并且引起利用反压力不能迫使通过的堵塞的趋势。这对于皂化混合物尤其适用。在进入步骤2后,物料足够稀使其没有问题。这种保持物料温热的问题也是可以看到的工艺问题的可能原因。无论何时单元式温度由于沸腾器故障所致下降太低时,重大和轻微的问题的频率上升。看到的大趋势之一是随着温度降低,水平读数变得更不规则。这可能是由于传感器的轻微淤塞。泵在较低温度下也变得不太可靠。这表明出现小的堵塞,其减慢或停止流动达一定时间段,直至所述泵最终使其通过。在步骤之间和在行动结束时,对整个系统冲洗并清洁。对于含水流点,运行热水以将大多数物料清洁掉。有机流点用庚烷或乙醇清洁。倾析器和karr柱二者的玻璃在不易于溶于水、乙醇或庚烷的材料的情况下快速淤塞。然而,发现流通蒸气除去大部分的所述材料。据信这是某种蜡并且蒸气熔化该物料并且水将其漂洗掉。一个非常有趣的发现在行动完成后,而清洁和清查正在进行时形成。在最周一周中较早取出并且在室温静置一周的一些酯化样品比预期更多地沉淀。通常酯化样品具有两相。顶相是在庚烷中的脂质,具有少量溶解的乙醇和酸。其颜色为非常深的褐色。底相通常是墨绿色层,其含有大多数的乙醇和酸,以及少量的水,和少量的固体。在静置一周之后,样品的顶层变亮至金色,非常类似于我们的最终产物看起来像的样子。对其的分析显示它仍然含有大多数EPA并且该EPA仍然为>97%EE。在底层中还有更多的固体。由此得出的理论是静置更长时间导致酸聚合一些重质“废弃物(junk)”或蜡并且它们变为某种烧焦的材料,然后从溶液中退出,留下浅颜色的油(图17)。综上所述,此行动非常成功。制得转化产物,其可以在精制中试工厂加工以制备最终65%EPA-EE产物。通过组合稳态数据,确定了连续转化工厂可以以97.5%转化率获得98%回收率,其相当于95.8%的收率。得到很多重要发现,其将对间歇或连续或可能一些混合形式的转化工厂的将来运行成形。补充数据图18和19很好地显示从进入塔顶馏出物流中的进料中完全回收脂质。进料和塔顶馏出物信号非常密切地匹配。尾分信号紧靠基线并且没有峰,意味着在尾分流中没有可测量的脂质。蓝色和红色信号没有完美对齐,因为它们来自转化程度不同的不同时间点。进料也没有如在这里看到的塔顶馏出物样品那样转化。其他Karr柱单元式操作称为水洗,因为它通过逆流地使水流动通过物料而从产物冲去乙醇和酸。对于此步骤的色谱图在图19中。进料和塔顶馏出物样品没有如在此Karr柱中那样很好地对齐。朝向行动结束,在GC中的注入口开始变脏。这导致峰在色谱图中轻微地移动。在这种情况下,塔顶馏出物比进料晚地运行,因此峰移动了一分钟的分数,使得峰没有完美对齐。尽管如此,仍然容易地看到信号看起来非常相似。而且,尾分信号几乎不可能看到,因为它紧靠基线而没有峰。这都显示所有的脂质从塔顶馏出物出来,并且在尾分中都没有损失,以及在此步骤期间没有发生化学变化。实施例4:初始乙醇反应工艺开发(反应1-6)A.概述进行实验以测试在我们的反应工艺中使用乙醇代替甲醇的似真性。必须调整的工艺的主要部分是反应后分离。因为乙醇在几乎所有中都混溶,所以液/液萃取不能如同在甲醇工艺中那样使用。对分离的第一次尝试利用己烷进行。将反应混合物过滤,然后直接放入转膜蒸发器(WFE)作为第二次尝试。还尝试水以迫使油进入单独的相。利用反应期间的蒸馏来除去大部分的乙醇以及导致较低转化率的水,这也有助于分离步骤。尝试不同的碱催化剂以判断有效性。这种工艺上的变化将去除对于酯交换步骤的需要并因此减少大量的投资和操作成本。而且,甲醇工艺使用都是有毒化学品的己烷和甲醇。在乙醇工艺中,完全去除甲醇。在一些实验中使用己烷,但最终除去。己烷的除去消除了对最终产品的污染问题之一。通过除去甲醇和己烷二者,所述工艺也对将来操作者具有更少的安全隐患。发现乙醇反应工艺使用乙醇汽提然后水洗涤反应器内容物而成功地从可能弄脏蒸馏步骤的一些其他废弃物分离脂肪酸乙酯产物。需要进行进一步的工作以更完全地定义该工艺。B.介绍到目前为止,反应工艺是既定步骤,其使用甲醇、酸和碱来将脂质官能化为甲酯。利用液液萃取,使用己烷来将酯从甘油和其他非所需化合物分离。在一些精制步骤之后,所述油随后将进行酯交换以使所有脂质形成乙酯,其是所需的终产物。在初始反应步骤中没有使用乙醇,因为它导致反应后液/液萃取的问题。C.实验和结果完成一系列反应,每次轻微改变步骤以试图确定如何得到所述油作为乙酯的完全转化和回收。1.反应1如同甲醇工艺一样运行反应,但是用200酒精度(proof)乙醇替代所述溶剂。将1514g乙醇萃取的粗品与3L的200酒精度乙醇和391g(3.5mol)的甲醇钾(KOMe)混合。理想地将使用乙醇盐,因为用酸中和所述碱将形成甲醇,其又可能形成甲酯,但是乙醇盐较慢到达,因此第一个反应使用甲醇盐。将混合物加热至沸点回流达一小时。此步骤称为皂化。在此期间,酸混合物通过将450ml的浓硫酸加入至1.5L的200酒精度乙醇构成。在皂化完成,并且反应器冷却低于沸点后,缓慢添加所述酸混合物,并使反应器回到沸腾达另外一小时。此步骤称为酯化。将混合物一分为二用于反应后处理实验。使用Buchner漏斗和1.5微米玻璃微纤维滤纸对两个半份进行过滤。一个半份进行旋转蒸发以除去溶剂,然后直接送至转膜蒸发器(WFE)以试图在没有初始液/液萃取的情况下蒸馏。第二个半份如在甲醇工艺中一样用己烷洗涤以促使甘油进入单独的相。然后将己烷在旋转蒸发器中脱去,接着通过WFE。己烷洗涤成功导致酯和甘油的分离。当分离的油通过WFE时,没有问题并且回收脂肪酸乙酯(FAEE)产物。直接来自WFE的产物为金黄色。在冰箱中静置一天后开始变为轻微的琥珀色,这是在来自甲醇工艺的最终乙酯产物中看到的。产物的所需颜色是金黄色。这显示颜色变化最可能是由于EFE而不是进一步下游的酯交换或蒸馏工艺所致。WFE可能开始这样的反应,其产生不期望的颜色并且在开始后,即使物料保持为冷,该反应也继续。在没有第一个分离步骤的情况下直接到WFE的混合物用黑硬物料堵塞WFE。经过一番讨论之后,确定这是残余硫酸与仍在混合物中的甘油和甘油类化合物反应而形成固体碳。对EPA与来自从己烷洗涤的WFE的乙酯形式的EPA(EPA-EE)进行质量平衡,确定反应回收率为34%。然而,由于所述混合物大致对半分,所以将接近于68%。2.反应2如同反应一一样运行反应2,直至酯化结束。将1504.3g粗品与在3L乙醇中的375.5g乙醇钠混合能够加热至回流达一小时。在皂化后加入在1.5L乙醇中的450ml硫酸,然后再次加热至大气压回流达一小时。在酯化后,将混合物冷却并使用乙醇钠中和。将混合物旋转蒸发以除去多余的溶剂,然后通过WFE以尝试分离。中和非常耗时。很难准确地计算所需的碱的量,所以其作为一个非常重复的过程结束。在另一个实验中,测试在乙醇在的pH测量的有效性,并且确定混合物需要具有至少20%的水以得到准确的pH。因此为了测量在添加碱之后的各个pH,选取一个样品并添加20体积%的DI水并且测量pH。然后添加更多的碱并重复该过程。反应器内容物上的最终pH为5.46。该中和总计用了369.2g。然后将混合物过滤。当将混合物放入WFE中时,其再次用黑色固体堵塞。因此,放弃将其直接放入WFE中的方法。3.反应3这是第一次尝试进行反应混合物的水洗以迫使油离开溶液。理论上油将漂浮至顶部并且水可以被拉到底部(除去)。甘油可溶于水但不溶于乙醇,所以它也将与水一起来到底部(除去)。然而,油和水都可混溶于乙醇,所以存在乙醇将充当阻止分离的共溶剂的问题。因此,相比于乙醇的量,需要的水量高以实现良好分离。还存在一个问题,即当pH仍然低时将水添加至混合物将导致逆反应至游离脂肪酸(FFA),因此降低工艺的效力。以与反应1和2的相同工艺通过酯化完成反应3。将1509.3g的粗品和溶解在3L乙醇中的375.7g的乙醇钠混合并加热至大气压回流达一小时。在皂化冷却之后添加在1.5L乙醇中的450ml硫酸。然后将反应器再次加热至大气压回流。在酯化之后,使用在水中的NaOH将混合物中和至pH为5.95。总体积为8L。向混合物中加入8L去离子(DI)水,然后将混合物过滤。使用1L乙醇来洗涤滤饼。过滤后的混合物使用20L分液漏斗分离。混合物没有良好分离,所以加入另外2L的DI水以进行酸分离。这次将其分类并且回收了80.9g的油产物。在将其在漏斗中静置较长时间之后,回收了另外的230.1g的油。这得到总计311g。关于EPA的质量平衡得到20%回收率。然而,考虑到至其他形式的脂质的乙酯的量,该反应将88%的EPA转化为乙酯形式,只是其没有良好回收。4.反应4此反应是试图获得己烷洗涤的反应的准确百分比回收率。将1507.3g的海藻粗品与375.7g的在3L乙醇中的乙醇钠混合。将混合物加热至大气压回流并保持一小时。通过缓慢将450ml的浓硫酸加入到1.5L的乙醇中制备酸溶液。在皂化步骤完成并且反应器冷却后,加入所述酸。然后将反应器重新加热至大气压回流达一小时。在酯化和冷却之后,将反应器内容物加入到具有8L己烷的20L大玻璃瓶(carboy)中。然后将该大玻璃瓶的内容物过滤,使用另外的8L己烷洗涤该大玻璃瓶和滤饼。将滤液置于分液漏斗中并静置30分钟。抛弃底层。具有油和己烷的顶层进行旋转蒸发。回收550g的产物油。对EPA的质量平衡得到以乙酯形式的58%回收率。可能一些EPA随底相离去。由于反应混合物完全溶于乙醇并且部分地溶于己烷,并且己烷和异常是混溶的,所以可能地分离是在己烷中可溶的物质和在己烷中不可溶的物质之间,其中脂质某种程度在这两个相之间分开。除去一些乙醇可能改善此分离。然而,己烷是有毒溶剂,所以它不是理想的。5.反应5在开始反应之前,使用旋转蒸发器干燥用于此反应的粗品。反应5以与反应2-4的相同方式通过酯化完成。大部分乙醇、连同生成的一些水使用简单蒸馏系统除去。在中和之前非故意地添加了水。计划是中和该反应然后进行水分离。据信由于脂质到游离脂肪酸的逆反应,这将使转化率变差,然而如通过以下数据显示的,情况并非如此。使用在水中的KOH完成中和。然后将混合物过滤并在分液漏斗中分离。对EPA进行质量平衡显示98%的回收率。6.反应6在旋转蒸发器中将用于此反应的粗品干燥并重建。以反应5的相同方式运行反应6,但是在中和之后而不是之前添加水。这是对于反应5的初始计划,但是由于操作者误差所致,必须进行另一个反应。此反应上的质量平衡显示比当在中和之前添加水时显著更低的61%的回收率。D.结果为了确定每个反应和分离步骤成功,测试起始粗品和最终FAEE产物的脂质含量。通过做质量平衡,计算%回收率。通过测试存在的乙酯相对于总脂质含量,计算%转化率。对于转化率,假定所有脂质在非极性相中而不是与水留在一起。以下是概述对于反应1-6的这些计算和数据的表。表:反应1-6的脂质数据和计算。基于该数据,反应5具有最佳回收率,并且所有的反应具有良好转化率。转化率部分意味着反应良好进行,回收率更多决定分离步骤如何好。由于甲基和乙酯之间的质量差异,最后两个反应的转化率超过%100。当测量总体脂质量时,脂质被官能化至甲酯。乙酯重量为约5%以上,并且因此105%的转化率是可能的。E.结论由于反应5的%回收率如此远高于其余的,所以那里遵循的在酯化后将乙醇汽提掉,然后在中和之前添加水的步骤适用于未来的反应。分离需要比初始预期更多的水。这可能是由于乙醇充当水和油之间的共溶剂。所以,可以脱去的乙醇越多,在较少水的情况下分离更好。这也可能导致己烷洗涤步骤中的较低回收率。尽管为了清楚理解的目的,前述发明已通过举例说明和实施例的方式在一些细节上进行了描述,但是本领域技术人员将理解某些改变和更改在所附权利要求的范围内可以实现。另外,本文中提供的每个参考文献以其整体结合,其程度如同每个参考文献单个地通过引用结合的程度相同。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1