一种基因组DNA的分析方法及其应用与流程
本发明属于基因修饰技术领域,具体涉及一种基因组dna的分析方法及其应用。
背景技术:
dna甲基化是真核细胞基因组重要修饰方式之一,在哺乳动物细胞的发育、分化、基因组印迹、以及基因表达调控等方面发挥了关键的作用,具有重要的生物学意义,并与人类疾病(包括肿瘤)的发生发展过程密切相关。因此,发现和建立与疾病过程相关的基因组dna修饰谱具有重要的应用价值和意义。
第一方面,dna甲基化通过影响染色体的构型和基因的转录,调控基因的功能。一般说来,dna甲基化与基因表达水平呈负相关关系,这不仅表现在基因启动子区的甲基化上,基因内部的甲基化水平也与基因表达负相关;反之,基因启动子区域的低甲基化与基因的转录活性正相关。体外研究表明,将高度甲基化的序列转入细胞后,基因表达受到抑制;而用甲基化抑制剂—5-叠氮胞嘧啶处理细胞,则能激活许多内源基因的表达。
第二方面,基因组dna甲基化具有调节胚胎发育的作用。基因组的甲基化模式在分化的体细胞中通常是稳定且可遗传的,但在哺乳动物生殖细胞的发育期和前胚胎期,通过大规模的去甲基化和再甲基化过程,形成新的甲基化模式,并产生有分化潜能的胚胎干细胞。
第三方面,基因组dna甲基化与基因组印记的形成和传递过程相关。尽管印记基因的形成机制还有待阐明,该现象的发生是源于等位基因的不同甲基化修饰而导致的结果,主要是cpg岛的胞嘧啶5’甲基化修饰。
第四方面,基因组dna甲基化与杂种优势的形成相关。利用甲级化敏感的多态性扩增方法(methylation-sensitiveamplifiedpolymorphism),研究者研究了亲本与杂种一代的甲基化差异对水稻杂种优势的影响。结果表明基因组的总体甲基化水平与杂种优势无关,而特定位点甲基化的改变则对杂种优势的形成具有显著效应。研究还发现,不同位点的甲基化对杂种优势的作用是不同的,有的对杂种优势的形成具有正效应,有的则为负效应。
第五方面,基因组dna甲基化水平与肿瘤的发生密切相关。基因组甲基化状态的改变是肿瘤发生的一个重要因素,这种变化包括基因组整体甲基化水平的降低和cpg岛局部甲基化水平的异常升高。这些变化会降低基因组的稳定性,如降低染色体稳定性、激活可移动遗传因子、启动原癌基因的表达、以及使抑癌基因失活等。因此,具体解析基因组dna甲基化修饰与肿瘤的关系,发现新的肿瘤标志物具有极其广泛的发展前景和应用价值。
1948年,科学家们第一次发现了dna的5-甲基脱氧胞苷,开启了基因组dna甲基化修饰研究的新篇章,相关研究衍生出三个主要的发展方向。第一个方向是研究基因组的整体甲基化水平,主要是将sssi甲基转移酶法、氯乙醛法等技术与高效液相色谱柱技术结合应用。第二个方向是位点特异的dna甲基化研究,使用的方法主要有甲基化敏感性限制性内切酶-pcr/southern法、重亚硫酸盐测序法、甲基化特异性pcr、甲基化敏感性单核苷酸引物延伸法、结合重亚硫酸盐的限制性内切酶法、甲基化敏感性单链构象分析法、甲基化敏感性变性梯度凝胶电泳法、甲基化敏感性解链曲线分析、methylight技术、dna微阵列法、甲基化敏感性斑点分析、ms-mlpa等技术方案。第三个方向是新的甲基化位点的检测,主要是应用限制性标记基因组扫描、mbd柱层法、以及compare-ms技术。这些方法为科学家进一步研究dna甲基化修饰提供了基础。
研究表明,dna甲基化与肿瘤的发生发展有着密切的联系。dna甲基化状态的改变包括高甲基化(hypermethylation)和低甲基化(hypomethylation)两种。一般来说,基因启动子区的dna高甲基化具有沉默基因表达的作用,而低甲基化则激活基因的表达。对不同肿瘤细胞的dna分析表明,癌变细胞中出现基因突变的概率要远远低于预期。而在转录组范围内检测结肠直肠癌中由启动子高甲基化引起的基因表达抑制,发现高达5%的已知基因在肿瘤细胞中发生了异常的启动子高甲基化。因此可以推测,与基因突变相比,dna甲基化改变在细胞恶变过程中可能发挥了更大的作用。
2015年之前,哺乳动物基因组dna的甲基化研究中仅发现了5-甲基脱氧胞苷修饰核苷,故上述与癌症相关的甲基化变化均为5mc。最近的研究报道,在果蝇、线虫的基因组dna中存在6ma修饰。2016年3月,《nature》杂志刊文报道了小鼠胚胎干细胞基因组dna中发现6ma修饰,首次确定了哺乳动物基因组dna中的一种新的修饰。科学家虽然6ma首次在哺乳动物体内报道,但是却未能阐述其功能。本发明利用hplc-ms/ms方法,检测细胞分化过程以及肿瘤细胞和正常细胞中5-甲基脱氧胞苷和n6-甲基脱氧腺苷的含量变化。阐述了n6-甲基脱氧腺苷的含量与细胞分化以及肿瘤发生的相关性,确立了一个新的肿瘤标志物-6ma。
技术实现要素:
本发明通过对多种肿瘤细胞与正常细胞、分化细胞与未分化细胞中n6-甲基脱氧腺苷(6ma)的含量变化研究,确立了将6ma作为新的肿瘤标志物以及检测6ma的方法。
首先,本发明提供了一种基因组dna的分析方法,该方法包括以下步骤:
(1)获得基因组dna;
(2)将步骤(1)中获得的基因组dna顺序与充分量的dnasei、nucleasep1以及牛肠碱性磷酸酶(calfintestinalalkalinephosphatase)接触,使长链dna分子充分降解为单核苷分子;其中,10×calfintestinalalkalinephosphatase缓冲液中含有1mnacl、0.5mnh4oac、0.1mmgcl2、及10mmdtt,使用浓氨水调节ph至8.2;
(3)将高效液相色谱和三重四级杆质谱联用,对步骤(2)获得的单核苷分子进行定性和定量分析,包括未修饰的和修饰的单核苷分子。
进一步地,本发明提供了一种基因组dna中6ma修饰核苷的分析方法,该方法包括以下步骤:
(1)获得基因组dna;
(2)将步骤(1)中获得的基因组dna顺序与充分量的dnasei、nucleasep1以及calfintestinalalkalinephosphatase接触,使长链dna分子充分降解为单核苷分子;其中,10×calfintestinalalkalinephosphatase缓冲液中含有1mnacl、0.5mnh4oac、0.1mmgcl2、及10mmdtt,使用浓氨水调节ph至8.2;
(3)将高效液相色谱和三重四级杆质谱联用,分析步骤(2)获得的单核苷分子;
(4)以6ma修饰核苷的标准品为参照,确定基因组dna中6ma修饰核苷的含量。在一种优选情况下,上述检测方法可用于检测基因组dna中6ma修饰核苷。
具体而言,上述检测方法可以是一种用于检测肿瘤细胞的方法,该方法包括如下步骤:
(1)获得待检测样品和正常对照样本的基因组dna;
(2)将步骤(1)获得的基因组dna顺序与充分量的dnasei、核酸酶p1以及牛肠碱性磷酸酶接触,使长链dna分子充分降解为单核苷分子;其中,10×calfintestinalalkalinephosphatase缓冲液中含有1mnacl、0.5mnh4oac、0.1mmgcl2、及10mmdtt,使用浓氨水调节ph至8.2;
(3)将高效液相色谱和三重四级杆质谱联用,定量检测基因组dna中6ma修饰核苷的含量;
(4)与正常对照样本相比,待检测样本中6ma含量小于等于67.85%为肝肿瘤细胞,小于等于50%为肺肿瘤细胞,小于等于54.55%为肾肿瘤细胞,小于等于46.67%为白血病患者的白细胞。
所述可以检测的肿瘤包括肝癌、肺癌、肾癌、及白血病。
具体而言,上述检测方法也可以是一种用于检测细胞或组织分化的方法,该方法包括如下步骤:
(1)获得待检测样品和对照样本未分化细胞的基因组dna;
(2)将步骤(1)获得的基因组dna顺序与充分量的dnasei、核酸酶p1以及牛肠碱性磷酸酶接触,使长链dna分子充分降解为单核苷分子;其中,10×calfintestinalalkalinephosphatase缓冲液中含有1mnacl、0.5mnh4oac、0.1mmgcl2、及10mmdtt,使用浓氨水调节ph至8.2;
(3)将高效液相色谱法和三重四级杆质谱联用,定量检测基因组dna中6ma修饰核苷的含量;
(4)与未分化细胞相比,待检测细胞中6ma的含量大于2.22倍时即为分化细胞。
一种促进细胞分化或逆转细胞癌变的方法,其特诊在于将细胞与能提高细胞基因组dna6ma水平的化合物接触。
本发明的有益效果
本发明提供了一种具有较高灵敏度和特异性的基因组dna分析方法,以及该方法在肿瘤检测中的应用。利用本发明提供的研究方法,我们发现6ma可作为一种肿瘤检测的标志物,并且通过本发明的基因组dna分析方法,可以定量检测基因组dna中6ma的含量。
附图说明
图1dna甲基化修饰的检测流程图
图2hplc检测基因组dna酶切效果
图3无tris引入的酶切基因组dna为单核苷的处理方法提高质谱灵敏度
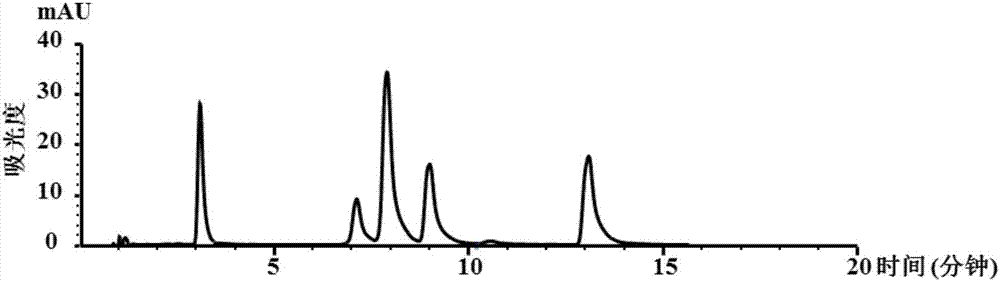
图4七种核苷的hplc-ms/ms检测图谱
图53t3-l1细胞分化过程中5-甲基脱氧胞苷和n6-甲基脱氧腺苷的检测
图6l-02、hepg2、mrc-5、a549细胞的5-甲基脱氧胞苷和n6-甲基脱氧腺苷的检测
图7人肿瘤组织和正常对照组织的5-甲基脱氧胞苷和n6-甲基脱氧腺苷的检测
图8hepg2细胞分化过程中5-甲基脱氧胞苷和n6-甲基脱氧腺苷的检测和定量
具体实施方式
下面结合具体实施例及附图,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如试剂盒说明书中所述的条件,或按照制造厂商所建议的条件。
实施例一、基因组dna的处理
采用北京博迈德基因技术公司的基因组dna提取试剂盒提取组织样本的基因组dna,采用天根生化科技公司的基因组提取试剂盒提取细胞基因组dna。将获得组织或细胞基因组dna进行rnasea处理,并使用天根公司的产物纯化试剂盒回收其中的基因组dna。,随后,使用dnasei、核酸酶p1(nucleasep1)、以及牛肠碱性磷酸酶(calfintestinalalkalinephosphatase)依次处理所获得的基因组dna,将其彻底降解为单核苷分子。该过程如图1所示。实验步骤具体如下:
基因组dna的处理:
1、缓冲溶液配制
在基因组dna处理过程中所使用的缓冲溶液包括20×rnaseabuffer、10×dnaseibuffer、5×nucleasep1buffer和10×rnaseabuffer,配方具体如下:
(1)20×rnaseabuffer
naoac6.8g
加水至49ml,使用冰醋酸调节ph至5.0,加水定容至50ml。保存在-30℃。
(2)10×dnaseibuffer
naoac3.4g
mgso40.253g
nacl0.07g
加水至49ml,使用2nhcl调节ph至5.0,加水定容至50ml。保存在-30℃。
(3)5×nucleasep1buffer
naoac82.0mg
zncl234.0mg
nacl146.3mg
加水至49ml,使用冰醋酸调节ph至5.3,加水定容至50ml。保存在-30℃。
(4)10×calfintestinalalkalinephosphatasebuffer
加水至49ml,使用浓氨水调节ph至8.2,加水定容至50ml。保存在-30℃。
2、组织或细胞基因组的提取
使用从北京博迈德基因技术有限公司购买的基因组dna提取试剂盒,提取组织基因组dna。使用从天根生化科技(北京)有限公司购买的细胞基因组提取试剂盒,提取细胞基因组。
3、rna去除
使用rnasea对提取的组织或细胞基因组进行处理,反应体系如下:
细胞或组织基因组100μl
20×rnasea5.6μl
buffer
2μg/μlrnasea溶5.6μl
液
将以上体系振荡混匀,瞬时离心后,37℃孵育12小时。
4、基因组dna的纯化回收。
使用从天根生化科技(北京)有限公司购买的大量产物纯化试剂盒,回收样本中的基因组dna。
5、dnasei酶切
使用dnasei,处理所提取的组织或细胞基因组dna,反应体系如下:
细胞或组织基因组dna30μg
10×dnaseibuffer5μl
dnasei60unit
加水补足至50μl。将以上体系振荡混匀,瞬时离心后,37℃孵育12小时。70℃孵育5分钟,冰浴5分钟。
6、核酸酶p1(nucleasep1)酶切
使用nucleasep1,进一步处理经dnasei处理后的dna样本,反应体系如下:
经过dnasei处理后的样本50μl
5×nucleasep1buffer15μl
nucleasep115unit
加水补足至75μl。将以上体系振荡混匀,瞬时离心后,37℃孵育12小时。
7、牛肠碱性磷酸酶(calfintestinalalkalinephosphatase)酶切
使用calfintestinalalkalinephosphatase,对经过nucleasep1处理后的样本做再次处理,反应体系如下:
经过nucleasep1处理后的样本75μl
10×calfintestinalalkaline10μl
phosphatasebuffer
calfintestinalalkalinephosphatase1unit
加水补足至100μl。将以上体系振荡混匀,瞬时离心后,37℃孵育12小时。
8、hplc检测酶切效果
使用agilent1260infinityquaternarylcsystem系统,对酶切产物进行分析。使用agilentzorbaxeclipseplusc18(4.6×100mm,3.5μm,agilent)色谱柱。水(a相)和甲醇(b相)作为流动相,流动相梯度为4min2.5-4.3%b,10.4min4.3-13.4%b,3.6min13.4-20%b,14min20-35%b,10min35%b,2min35-95%b,11min95%b,1min95-2.5%b,10min2.5%b。实验结果如图2所示,表明利用本实施例建立的基因组dna处理方案,能够将基因组dna完全降解为单核苷酸分子,为定性和定量分析分子其中的修饰核苷奠定了基础。
10×calfintestinalalkalinephosphatase缓冲液的配制:向49ml双蒸水中,加入2.93gnacl(终浓度1m),1.93gnh4oac(终浓度0.5m),0.48gmgcl2(终浓度0.1m),77.1mgdtt(终浓度10mm),用浓氨水将溶液ph调节至8.2,加水定容至50ml。与文献报道不同,我们使用浓氨水代替tris调节ph,避免了tris对离子源的不利影响,提高了检测灵敏度,使检测最低限降低了100倍。
实施例二、dna甲基化修饰的检测和定量
利用冷冻离心浓缩仪,将获得的核苷分子样本进行浓缩至干,使用乙腈脱盐后,用30μl水复溶。选用atlantist3柱,用shimadzulc-20ahplcsystem和absciexqtrap5500组成的hplc-ms/ms对样品溶液进行检测。具体操作步骤如下:
1、获得经过实施例一中所述方法处理的基因组dna的单核苷样本;
2、将步骤(1)中获得的单核苷样本40℃,3000rpm,0.01mpa,360min进行离心浓缩至干;
3、将步骤(2)中获得的单核苷样本用100ul乙腈复溶、12000rpm离心10min脱盐,吸取上清;
4、将步骤(3)中获得的单核苷样本的乙腈溶液40℃,3000rpm,0.01mpa,120min进行离心浓缩至干;
5、将步骤(4)中获得的单核苷样本用100ul高纯水复溶,4℃静置24h;
6、将步骤(5)中获得的单核苷样本水溶液加入同等体积的1ug/ml的6-氯嘌呤;
7、将步骤(6)中获得的样本水溶液通过上述基因组dna甲基化修饰的分析方法进行种类和含量的分析。
分析条件:
1、流动相梯度:
3mm醋酸铵作为a相,甲醇作为b相,流动相梯度如下:
2min2%b,4min2-5%b,2min5-10%b,4min10%b,0.1min10-95%b,2min95%b,0.1min95-2%b,4.9min2%b。
2、离子源参数
3、ms/ms参数
实验结果如图3、图4所示。上述实验结果表明,利用本实施例建立的技术方案,能够准确的检测细胞或组织基因组dna中的甲基化修饰。使用浓氨水代替tris调节ph,避免了tris对离子源的掩蔽作用。与tris处理的样本相比,提高了检测灵敏度,使检测限降低了100倍。
实施例三、3t3-l1细胞分化过程中,甲基化修饰的检测和定量
使用人重组胰岛素、地塞米松和3-异丁基-1-甲基黄嘌呤,诱导3t3-l1细胞进行脂肪细胞分化。具体步骤如下:
1、在实验的第0天,进行3t3-l1细胞铺板。在每个10cm培养皿,加入275×104~330×104个3t3-l1细胞;
2、48小时后,细胞生长达到完全融合状态;
3、加入含有10%fcs,1mg/ml人重组胰岛素,0.5mmol/l3-异丁基-1-甲基黄嘌呤,1μmol/l地塞米松的dmem培养基,对3t3-l1细胞进行诱导分化;
4、48小时后,使用含有10%fcs和1mg/ml人重组胰岛素的dmem培养基替换步骤3中的培养基;
5、从实验的第六天起,使用含有10%fcs的dmem培养基替换步骤4中的培养基。每两天更换一次。
使用利用实施例一和实施例二所述方法,对3t3-l1细胞分化过程中第0天,第2天,第3天,第4天和第6天的甲基化修饰进行检测。通过计算5mc甲基化修饰碱基数目占基因组dna碱基总数目的百分比,获得5mc甲基化修饰碱基的含量。通过计算6ma甲基化修饰碱基数目占基因组dna碱基总数目的百分比,获得6ma甲基化修饰碱基的含量。
实验结果如图5所示。研究结果表明,分化过程中,3t3-l1细胞细胞基因组dna中的甲基化修饰含量呈现明显变化,随着分化过程的加强,5mc和6ma甲基化修饰核苷的含量显著提高。这种情况表明,5mc和6ma甲基化修饰核苷的含量与细胞的分化状态密切相关,细胞分化程度越低,5mc和6ma甲基化修饰核苷的含量越低。其中,未分化细胞的5mc甲基化修饰含量为0.35135%-0.58594%,6ma甲基化修饰含量为0.00206%-0.01637%,分化细胞的5mc甲基化修饰含量为0.72414%-0.96272%,6ma甲基化修饰含量为0.00997%-0.07247%。与未分化细胞相比,分化细胞的6ma甲基化修饰碱基的含量提高了2.2倍,5mc甲基化修饰碱基的含量提高了1.5倍。
实施例四、定性和定量检测l-02、hepg2、mrc-5和a549细胞中dna的甲基化修饰
使用利用实施例一和实施例二所述方法,对l-02(正常肝细胞)、hepg2(肿瘤肝细胞)、mrc-5(正常肺细胞)、a549(肿瘤肺细胞)细胞的甲基化修饰进行检测,实验结果如图6所示。
研究结果表明,l-02细胞基因组dna中的6ma甲基化修饰含量为hepg2细胞的3.7倍,mrc-5细胞基因组dna中的6ma甲基化修饰含量为a549细胞的6.1倍,均存在显著性差异。表明与正常细胞相比,肝癌和肺癌肿瘤细胞基因组中的6ma甲基化修饰核苷的含量均出现显著降低。对于5mc甲基化修饰而言,l-02细胞与hepg2细胞基因组dna中的5mc甲基化修饰含量基本持平;a549细胞比mrc-5细胞的5mc含量较低,但其间方差异不具有显著性。上述结果表明,与正常细胞相比,肿瘤细胞基因组dna中6ma甲基化修饰碱基的含量显著降低,这与细胞分化过程中6ma甲基化修饰碱基含量的变化是一致的。这个结果也提示我们,6ma可以作为一种肿瘤检测的标志物。
实施例五、定性和定量检测肿瘤和癌旁组织中dna的甲基化修饰
获得经手术分离的肝癌、肺癌、肾癌组织及对应的癌旁组织样本,获得白血病患者及健康对照的白细胞,使用利用实施例一和实施例二所述方法,对癌组织和癌旁组织/健康对照的甲基化修饰进行检测,实验结果如图7所示。
研究结果表明,与正常组织相比,肿瘤组织基因组中6ma甲基化修饰核苷的含量显著降低,而5mc修饰碱基含量的变化不太明显。具体而言,肝正常组织基因组dna中的6ma甲基化修饰含量为2.214×10-4%-2.809×10-4%,肝肿瘤组织基因组dna中的6ma甲基化修饰含量为0.3926×10-4%-1.641×10-4%;肺正常组织基因组dna中的6ma甲基化修饰含量为2.904×10-4%-3.429×10-4%,肺肿瘤组织基因组dna中的6ma甲基化修饰含量为0.6198×10-4%-1.280×10-4%;肾正常组织基因组dna中的6ma甲基化修饰含量为2.769×10-4%-3.198×10-4%,肾肿瘤组织基因组dna中的6ma甲基化修饰含量为0.6000×10-4%-1.433×10-4%;正常人群白细胞基因组dna中的6ma甲基化修饰含量为1.410×10-4%-2.018×10-4%,白血病患者白细胞基因组dna中的6ma甲基化修饰含量为0.4899×10-4%-0.7387×10-4%。
与癌旁对照组织相比,肝癌组织基因组dna中6ma甲基化修饰的含量为癌旁组织的67.85%以下,肺癌组织基因组dna中6ma甲基化修饰的含量为癌旁组织的50.00%以下,肾癌组织基因组dna中6ma甲基化修饰的含量为癌旁组织的54.55%以下。以健康人的白细胞为正常对照,白血病患者白细胞基因组中6ma甲基化修饰的含量为正常对照的46.67%以下,且四组数据均存在显著性差异。
临床样本的研究结果显示,6ma可以作为一种肿瘤检测的标志物。
实施例六、hepg2细胞诱导分化过程中dna甲基化修饰的检测和定量
使用全反式维甲酸诱导hepg2细胞分化。具体步骤如下:
1、用二甲基亚砜(dmso)将全反式维甲酸配制成10mmol/l溶液,以1mol/lnaoh调ph至7.0,过滤后分装,置于-20℃保存;
2、在实验的第0天,进行hepg2细胞铺板。每个10cm培养皿,铺275×104~330×104个hepg2细胞。
3、将细胞培养物分为两组,第一组加入含有0.1%dmso的培养基,第二组加入含有10μmol/l全反式维甲酸的培养基。
4、每两天换液一次,持续培养。
使用利用实施例一和实施例二所述方法,对hepg2细胞分化过程中第0天,第1天,第2天,第4天,第7天和第14天的甲基化修饰进行检测。实验结果如图8所示。
上述结果表明,在hepg2细胞体外诱导分化过程中,基因组dna中5mc和6ma甲基化修饰碱基的含量显著升高。诱导分化的第14天,hepg2细胞基因组中6ma甲基化修饰碱基的含量为第0天的4.4倍,5mc甲基化修饰碱基的含量为第0天的1.2倍。t检验显示第14天和第0天的6ma含量与5mc含量均具有显著性差异,friedman检验证明诱导分化过程中,6ma含量变化、5mc含量变化均具有显著性差异。
上述结果表明,hepg2细胞体外诱导分化过程中,基因组dna中6ma甲基化修饰碱基的含量出现显著变化。这个结果也表明,6ma与肿瘤诱导分化相关,并可以作为肿瘤治疗的一个靶点。
- 还没有人留言评论。精彩留言会获得点赞!