一种泊沙康唑的合成方法与流程
本发明涉及药物合成领域,一种泊沙康唑的合成方法。
背景技术:
泊沙康唑(中文化学名:4-[4-[4-[4-[[(3R,5R)-5-(2,4-二氟苯基)-5-(1,2,4-三唑-1-基甲基)氧杂戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基]-2-[(2S,3S)-2-羟基戊-3-基]-1,2,4-三唑-3-酮),具有下式I所示结构。
泊沙康唑是由德国先灵葆雅公司研制,商品名“Noxafil”,于2005年10月20日在欧盟和美国FDA获得批准上市的抗真菌药物。泊沙康唑口服混悬液于2006年9月再次获美国FDA批准用于骨髓移植后免疫力减弱和癌症化疗引起白细胞减少的患者,以防止曲霉菌和念珠菌引起的感染。由于其具有高效低毒的特点,且临床应用范围广,疗效较好,尤其是对多烯类化合物和其他三唑类耐药或侵袭性真菌感染有效,为临床侵袭性真菌感染的治疗提供了选择空间。
现有技术中关于泊沙康唑的合成方法已有大量研究,一般最终均由如下式II所示化合物氢化脱苄基得到泊沙康唑。
US5625064和CN101824009A中记载:将式II所示化合物冷却至50℃,加入350ml甲酸,继续冷却至20℃,加入另外350ml甲醇,取85gPd/C加入350ml甲酸搅拌成浆状加入反应瓶中,在20℃下搅拌过夜,加热至40℃反应24h,抽滤,滤饼用350ml甲酸、700ml甲醇淋洗。减压蒸干溶剂,向残余物中加入3500ml甲醇和700ml氨水,加热至回流1-2h,然后冷却析出大量固体,抽滤,滤饼用1400ml甲醇和水(1:1)洗涤,40℃下鼓风烦躁得到300.5g终产物。
Anil K.Saksena(Tetrahedron Letters 45(2004)8249–8251)指出:将4.03g式II所述化合物溶于70ml甲酸中,再加入5%Pd/C 8g,反应混合物在20℃下搅拌15h,然后加热至35-40℃,再搅拌24h。然后将反应液降温至20℃,过滤,并用约40ml甲酸洗涤滤饼,再用35ml甲醇洗涤滤饼两次,滤液旋蒸至干得剩余物,柱层析,得泊沙康唑3g,收率为84%。
上述工艺方法中泊沙康唑的合成路线如下
在以上两种方式中,甲酸即为反应溶剂,也是氢源。先在20°下反应,再升高温度至35-40℃反应,总反应时间均超过40h,且后处理需柱层析纯化样品,溶剂量太大,不适应于大生产。
在CN102863431A中记载:250ml三口瓶中加入式II所示化合物(8.12g)和氢溴酸70ml,搅拌至固体溶解,加热至50℃反应2-3h,液相监测反应完毕。反应液用水(100ml)稀释,NaOH水溶液(100ml,25%)调节至pH约为9,二氯甲烷(100ml)萃取,有机相10%NaOH水溶液(100ml)洗涤,水(100ml)洗涤,饱和NaCl水溶液(100ml)洗涤,无水NaSO4干燥,浓缩,真空干燥,得到土黄色粗产物,纯度为93.9%。该实验方法中,氢溴酸即为反应溶剂也为氢源,其具有强烈的棘刺味道且不易储存,易被氧化。且后处理繁琐,收率低,不适合应用于大生产。
此外,在WO2013042138A2和US2014343285A1中记载:在420ml甲醇中加入42g式II所示化合物,5mol/l盐酸和10%Pd-C,在4-5kg/cm2(0.4-0.5Mpa)氢气压力,反应温度50℃下反应5h,反应结束后,过滤除去催化剂,然后用4mol/L氢氧化钠溶液调节ph值为7左右,加入水并在25-35℃下搅拌2h,有大量固体析出,过滤,并用水洗涤滤饼,滤饼在异丙醇中重结晶得到泊沙康唑,收率为75%,纯度为99.85%。该工艺方法中泊沙康唑的合成路线如下:
上述实验方案虽然在一定程度上缩短了反应时间,提高了标产物的纯度。然而为了能够实现泊沙康唑的大规模工业化生产,对于泊沙康唑的合成方法还有待于进一步优化和改进,以建立了一种反应时间更短、且泊沙康唑的纯度和收率均有所提高的泊沙康唑的合成方法。
技术实现要素:
本发明的主要目的在于提供一种泊沙康唑的合成方法,以在提高泊沙康唑的收率和纯度的同时,提供一种适用于大规模工业化生产的泊沙康唑的合成方法。
为此,在本发明中提供了一种泊沙康唑的合成方法,该合成方法包括下列步骤:
S1、以如下式II所示化合物为原料,以氢气为氢源,在氢化脱苄基反应条件下,将式II所示化合物与催化剂和酸性试剂在反应溶剂中搅拌接触反应,得到混合溶液;
S2、将所述混合溶液降温至0-5℃,向所述混合溶液中滴加氢氧化钠溶液至混合溶液的pH值为10-12,搅拌过滤得滤饼,清洗后干燥得到如下式I所示的泊沙康唑。
根据本发明的合成方法,添加催化剂的目的主要是促使氢化脱苄基反应发生,其中对于催化剂的使用并没有特殊要求,可以采用本领域能够促使氢化脱苄基反应发生的任意的催化剂。然而为了优化反应效果,提高泊沙康唑的收率,优选以含水量为50wt%的10%钯碳催化剂作为催化剂(其中10%钯碳催化剂为钯含量为催化剂干基重量的10wt%的催化剂),且所述10wt%钯碳催化剂的干重与式II所示化合物的重量比为(0.05-0.1):1,优选为(0.08-1):1,更优选为0.08:1。
根据本发明的合成方法,添加酸性试剂的主要目的为了使式II溶解于反应溶剂中,其中对于酸性试剂的使用并没有特殊要求,可以是本领域常规的任意能够满足上述目的的有机酸或无机酸。然而为了优化反应效果,提高泊沙康唑的收率,优选所述酸性试剂与式II所示化合物的体积重量比为(0.5-1.5)L:1kg,更优选为1L:1kg。
优选地,所述酸性试剂为盐酸、硫酸,冰醋酸、甲酸和苯磺酸中的一种或多种,更优选所述酸性试剂为盐酸。在本发明中对于这些酸性试剂的浓度并没有特殊要求,可以根据实际需要调整。
根据本发明的合成方法,重点在于以式II所示化合物为原料,以氢气为氢源在氢化脱苄基反应条件下进行搅拌反应,进而在加快反应效率的同时,提高泊沙康唑的收率,并简化后处理步骤。在本发明中对于步骤S1中氢化脱苄基反应条件并没有特殊要求,可以参照常规的合成方法中的相应条件,在本发明中优选所述氢化脱苄基反应条件包括:在常压、温度为50-55℃条件下,以350-450rpm的速度,搅拌反应2-3h。
根据本发明的合成方法,为了满足步骤S1中搅拌反应的要求,优选情况下,促使步骤S1在内置有搅拌器的反应釜中进行。这种内置有搅拌器的反应釜可以采用常规市售的反应釜。然而为了优化搅拌效果,促使物料在搅拌的情况下进行传热、传质和反应,进而提高反应速率,在一种优选实施方式中,所述反应釜中搅拌器包括搅拌轴和沿所述搅拌轴的轴线方向均布的多组搅拌叶片组,各个搅拌叶片组中分别包括沿所述搅拌轴周向均布的1至3片搅拌叶片。优选地,相邻两组搅拌叶片组中搅拌叶片平行排布或者交错排布。优选的所述搅拌叶片沿所述反应釜径向的长度为所述搅拌釜半径的1/5-2/5倍,优选为1/3倍。
根据本发明的合成方法,为了进一步优化搅拌效果,促使反应加速,优选所述反应釜中还设有至少一组挡板组,所述一组挡板组中包括多个挡板、且各所述挡板的长度方向沿平行于所述搅拌器的轴线方向设置、宽度方向沿所述反应釜的径向方向设置。优选所述搅拌器沿所述反应釜的轴线设置,且所述一组挡板组中各所述挡板以所述搅拌器的轴线为垂直中分线呈圆周分布。优选的各所述挡板的高度为搅拌釜高度的1/2-2/3倍,宽度为所述搅拌釜半径的1/6-1/4倍,厚度为0.5-1cm。通过在搅拌釜中设置挡板组,促使被搅拌器搅动的液体与挡板发生冲击,促进式II所示化合物与氢气、催化剂、以及酸性试剂在搅拌的情况下进行传热、传质和反应,进而提高反应效率。
在一种优选实施方式中,所述反应釜中设有一组挡板组,且该挡板组中各挡板沿其宽度延伸的方向,一侧固定在所述反应釜的内壁上,另一侧朝向所述搅拌器的位置延伸。
在另一种优选实施方式中,所述反应釜中设置两组或三组挡板组,各所述挡板组以所述搅拌器的轴线为垂直中分线呈同心圆设置、且相邻两组挡板组中的挡板交错设置。
根据本发明的合成方法,在步骤S2中将所述混合溶液降温至0-5℃,在该步骤中对于所选择的降温方式并没有特殊要求,在本发明中优选直接在混合溶液中加入冰块以进行快速降温,这种快速降温能够减少副反应的发生。
在一种优选的实施方式中,根据本发明的合成方法包括以下步骤:S1、以如下式II所示化合物为原料,以氢气为氢源,将式II所示化合物、10%钯碳催化剂和酸性试剂按比例(具体比例参照前述说明,在此不再赘述)在反应釜中搅拌混合,在常压、50-55℃温度下,以350-450rpm的速度,搅拌反应2-3h(HPLC监测反应,控制反应终点),分离催化剂后得到混合溶液;其中所述反应釜为内置有搅拌器和任选的至少一组挡板组的反应釜(所述搅拌器和所述挡板组的结构如前所述在此不再赘述);S2、将所述混合溶液降温至0-5℃,向所述混合溶液中滴加碱性试剂至混合溶液的pH值为10-12,搅拌过滤得滤饼,清洗后干燥得到如下式I所示的泊沙康唑。
根据本发明的合成方法,其中对于获取式II所示化合物的方法并没有特殊要求,其按照现有技术中的常规方法合成即可,例如:在100ml三口瓶中加入2-[(1S,2S)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3H-1,2,4-三氮唑-3-酮(4.78g)和DMSO(40ml),搅拌溶解,加入浓度25wt%的NaOH水溶液(1.6ml),混合搅拌10分钟,加入(5R-CIS)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1H-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(4.4g),混合物30℃反应约14h,液相监测至反应完毕,将反应液倒入剧烈搅拌的水(240ml)中,加毕,再剧烈搅拌10min,过滤,滤饼用水洗涤,将滤饼用异丙醇重结晶后得到固体,真空干燥得到灰白色粉末固体(式II)。
同时,在本发明中还提供了一种由本发明所述的方法合成的泊沙康唑,优选所述泊沙康唑的纯度≥99%。
根据本发明所提供的泊沙康唑的合成方法以式II所示化合物为原料,以氢气为氢源,在搅拌条件下促使式II所示化合物发生氢化脱苄基反应。该方法不但降低了反应对于压力的要求,而且明显缩短了反应时间,只需2-3h即可;而且后处理简便,低温下调节ph值至10-12,离心,即可得到目标产物泊沙康唑,该方法适用于大规模的工业化生产。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
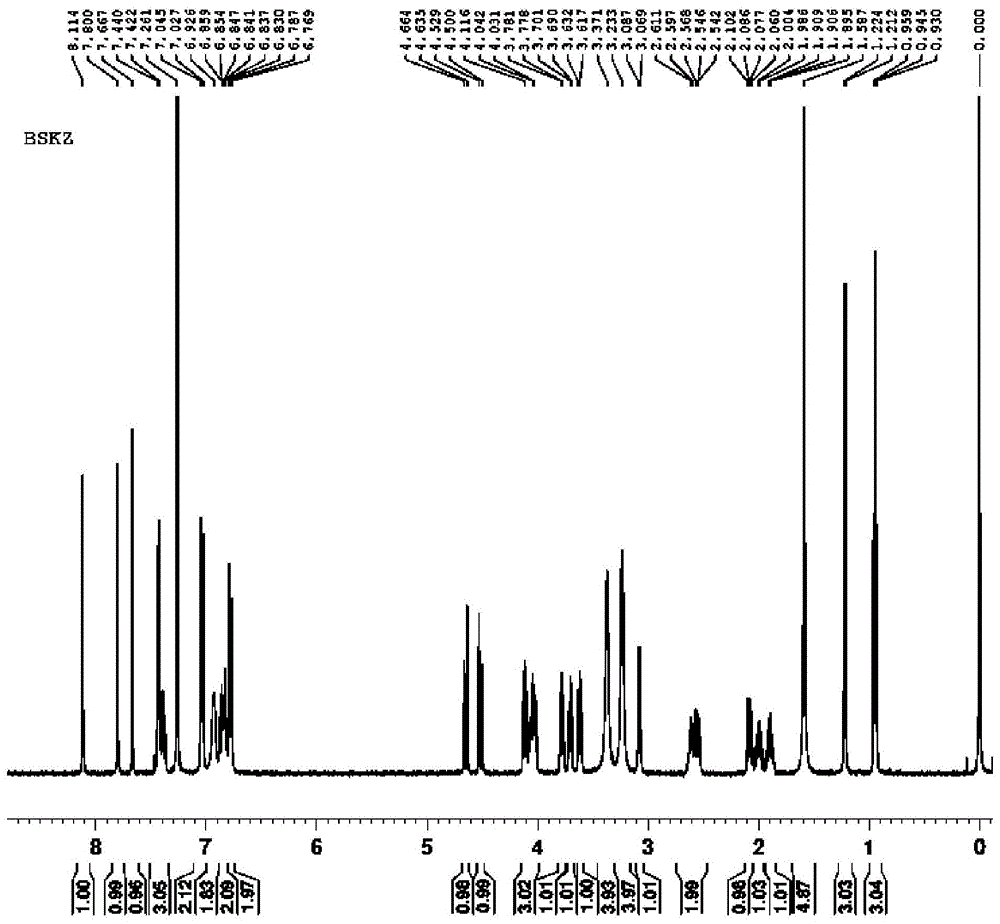
图1为实施例1所合成的泊沙康唑的核磁氢谱图;
图2为实施例1所合成的泊沙康唑的核磁碳谱图。
具体实施方式
在实施例1至4中所采用的式II所示结构的化合物的合成方法如下:
在100ml三口瓶中加入2-[(1S,2S)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3H-1,2,4-三氮唑-3-酮(4.78g)和DMSO(40ml),搅拌溶解,加入浓度25wt%的NaOH水溶液(1.6ml),混合搅拌10分钟,加入(5R-CIS)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1H-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(4.4g),混合物30℃反应约14h,液相监测至反应完毕,将反应液倒入剧烈搅拌的水(240ml)中,加毕,再剧烈搅拌10min,过滤,滤饼用水洗涤,将滤饼用异丙醇重结晶后得到固体,真空干燥得到灰白色粉末固体(式II);经核磁、质谱以及红外光谱综合检测可知,该灰色粉末固体为式II所述化合物,且该化合物的纯度为>99%。
实施例1
用于说明本发明泊沙康唑及其的合成方法
(1)内置有搅拌器的反应釜结构:
该反应釜的高度为120cm,直径为50cm的100L搪瓷反应釜。其中设有搅拌器和一组挡板组,所述搅拌器为标配,包括沿所述反应釜的轴线方向设置的搅拌轴和沿所述搅拌轴的轴线方向均布的两组组搅拌叶片组,各组搅拌叶片组平行设置,且每组搅拌叶片组中包括两片沿所述搅拌轴周向均布的搅拌叶片,每片搅拌叶片沿所述反应釜径向方向的长度8cm;所述挡板组包括高度为60cm,宽度为5cm,厚度为1cm的4个挡板,各挡板的长度方向沿平行于所述搅拌器的轴线方向设置(一侧固定在所述反应釜的底壁上)、宽度方向沿所述反应釜的径向方向设置,各挡板沿其宽度延伸的方向(一侧固定在所述反应釜的侧壁上,另一侧朝向所述搅拌器的位置延伸),且各挡板以所述搅拌器的轴线为垂直中分线呈圆周分布。
(2)泊沙康唑的合成
在上述100L搪瓷反应釜中加入60L甲醇,4kg式II所示化合物,4L浓度为36.5wt%的盐酸,320g的10%钯碳催化剂(含水50%),密封反应釜,开启搅拌器(搅拌速度为350rpm)。先用氮气置换釜内空气3次,再用氢气置换3次,氢气压力保持在0.1MPa(表压),升温加热至50℃,HPLC监测反应,2h反应结束。降温,用氮气置换3次,过滤得到10%钯碳催化剂。将滤液倒入另一个200L搪瓷反应釜中,加入冰块,降温至0-5℃,向滤液中滴加2mol/L NaOH溶液,调节pH至11,有大量固体析出,搅拌2h后,离心,并用水洗涤滤饼3次,将固体放于55℃鼓风干燥箱中干燥,得到3.32kg的产物,附图1示出了该产物的核磁氢谱图,附图2示出了该产物的核磁碳谱图,结合附图1和2可知,该产物正是所需泊沙康唑,经计算该产物的收率为93.6%,HPLC纯度为99.754%。
实施例2
用于说明本发明泊沙康唑及其的合成方法
(1)内置有搅拌器的反应釜结构:同实施例1。
(2)泊沙康唑的合成:
在前述100L的搪瓷反应釜中加入,加入60L甲醇,4kg式II所示化合物,6L浓度为98wt%的硫酸,400g的10%钯碳催化剂(含水50%),密封反应釜,开启搅拌器(搅拌速度为400rpm)。先用氮气置换釜内空气3次,再用氢气置换3次,氢气压力保持在0.1MPa(表压),升温加热至55℃,HPLC监测反应,2h反应结束。降温,用氮气置换3次,过滤得到10%钯碳催化剂。将滤液倒入另一个200L搪瓷反应釜中,加入冰块,降温至0-5℃,向滤液中滴加2mol/L NaOH溶液,调节pH至12,有大量固体析出,搅拌2h后,离心,并用水洗涤滤饼3次,将固体放于55℃鼓风干燥箱中干燥,得到3.27kg的产物,通过核磁氢谱图核磁碳谱图可知,该产物正是所需泊沙康唑,经计算该产物的收率为92.3%,HPLC纯度为99.594wt%。
实施例3
用于说明本发明泊沙康唑及其的合成方法
(1)内置有搅拌器的反应釜结构:同实施例1;
(2)泊沙康唑的合成:
在前述100L的搪瓷反应釜中加入,加入60L甲醇,4kg式II所示化合物,4L浓度为36.5wt%的盐酸,200g的10%钯碳催化剂(含水50%),密封反应釜,开启搅拌器(搅拌速度为450rpm)。先用氮气置换釜内空气3次,再用氢气置换3次,氢气压力保持在0.12MPa,升温加热至50℃,HPLC监测反应,3h反应结束。降温,用氮气置换3次,过滤得到10%钯碳催化剂(备用)。将滤液倒入另一个200L搪瓷反应釜中,加入冰块,降温至0-5℃,向滤液中滴加2mol/L Na2CO3溶液,调节pH至10,有大量固体析出,搅拌2h后,离心,并用水洗涤滤饼3次,将固体放于55℃鼓风干燥箱中干燥,得到3.24kg的产物,通过核磁氢谱图核磁碳谱图可知,该产物正是所需泊沙康唑,经计算该产物的收率为91.4%,HPLC纯度为99.125%。
实施例4
用于说明本发明泊沙康唑及其的合成方法
(1)内置有搅拌器的反应釜结构:该反应釜的高度为120cm,直径为50cm的100L搪瓷反应釜。其中设有搅拌器(未设挡板),所述搅拌器包括沿所述反应釜的轴线方向设置的搅拌轴和沿所述搅拌轴的轴线方向均布的四组搅拌叶片组,各组搅拌叶片组平行设置,且每组搅拌叶片组中包括3片沿所述搅拌轴周向均布的搅拌叶片,搅拌叶片沿所述反应釜径向方向的长度8cm。
(2)泊沙康唑的合成:参照实施例中泊沙康唑的合成,经反应得到3.19kg的产物,通过核磁氢谱图核磁碳谱图可知,该产物正是所需泊沙康唑,经计算该产物的收率为90.1%,HPLC纯度为98.565%。
从上述实施例的结果中能够看出,利用本发明的方法合成式(I)的化合物简便易行,而且反应效率高且产率高。而且,如上文所述,US5625064和CN101824009A中报道技术方法需要反应40h;WO2013042138A2和US2014343285A1中报道技术虽反应时间为5h,但目标产物经过异丙醇重结晶后纯度才达到99.85%;CN102863431A中报道技术方法的后处理太繁琐,且纯度太低(93.9%)。本发明通过以氢气为氢源,在搅拌条件(设置搅拌器和任选的挡板)下完成氢化脱苄基反应,明显缩短了反应时间(2-3h),且后处理简便,纯度高(>98.5%)。本发明的方法克服了现有技术的托法替布合成方法中的合成工艺过程复杂,生产周期长的缺陷,适合于大规模工业化生产。而且,在合成泊沙康唑的后续处理工艺中,利用本发明所述方法合成能够减少杂质的引入,有利于在药学领域的应用。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!