一种给受体型芴基纳米格子材料、制备方法及其应用与流程
本发明属于有机半导体材料技术领域,具体涉及可溶液加工的给受体型具有纳米级孔径的给受体型芴基纳米格子材料及其制备方法,并涉及这些材料在有机电存储、有机电致发光、光伏电池、光探测、化学与生物传感和有机激光等领域的应用。
背景技术:
有机光电器件由于生产成本低、制作工艺简单、可制备柔性大面积器件等优点,业已成为目前国内外科学工作者研究的热点。有机小分子材料由于分子结构确定,易提纯等优点已广泛应用于有机太阳能电池、有机电致发光、有机光探测、有机存储、有机激光等领域。自1993年,H.Wynberg等[Havinga1 E.E.,Hoeve W.ten,Wynberg H..Synthetic Metals,1993,299]提出电子给体-电子受体的分子设计和1998年黄维等[Yu W-L,Meng H,Pei J,Huang W,Li Y,Heeger A J.Macromolecules,1998,31,4838]将给受体(p-n)型的设计应用于制备有机功能材料以来,此设计已成为目前有机功能材料的主流。给受体型分子设计通过电子给体、电子受体的模块化设计与组合可以实现对有机分子的带隙、能级排布的精确调控。而对有机材料的电学带隙、能级排布、载流子迁移率等有效的调控是提高有机光电器件性能的关键。
到目前为止,大环状化合物一般分为两类:一类是如COFs、MOFs等大环化合物,但是刚性的骨架使其具有独特物理化学性质,诸如结构稳定性好、空间尺寸可调、比表面积大、介电常数低等在气体吸附、化学分离、多相催化等领域展现良好的应用前景。部分结构由于紧密的π-π堆积,相对于一般有机物展现出优异的载流子迁移率,在光电器件领域有潜在的应用前景,但是不能溶液加工限制了其进一步应用于光电器件。另一类主要以近年来刚开始研究的新型环状化合物如柱芳烃类(Pillararene)、凯库勒烯类(kekulene)、[n]环对苯撑([n]CPPs)以及类石墨烯或碳纳米管类环状小分子或者聚合物,此类化合物具有刚性的骨架结构兼具可溶性加工的优点,在光电材料领域初步展现出迷人的应用前景,但是此类可柔性加工的材料鲜有骨架中同时含有给受体片段交替排列的分子构型报道,受体片段的引入通常是通过大环骨架桥连实现,主要是由于环状分子成环产率低,难以分离、受体片段的反应活性低等原因难以实现。因而无法实现对环状化合物带隙、能级等光电性质参数的有效调控。同时,环状化合物纳米级孔径的精确调控、高性能光电基团的引入等也面临一系列挑战,材料光电领域的应用研究刚刚起步,不够深入。
技术实现要素:
发明目的:鉴于以上具有纳米级尺寸格子分子目前存在的问题,本发明公开了一类给受体型具有纳米级孔径的环状格子半导体材料及其制备方法。此类分子成功将电子受体单元引入环状分子的骨架,合成步骤简单实用,成环前体通过一步Friedel–Crafts自身缩合反应得到,综合产率高。成环前体可拓展性好,从而可实现丰富官能团的引入及纳米级孔径的精确调控。给受体单元同时引入格子分子骨架可实现化合物带隙、能级等光电性质参数的精确调控。本发明公开的给受体型芴基衍生物纳米格子分子良好的溶解性以及热、电化学和光谱稳定性兼具刚性的骨架,是一类非常具有应用前景的有机光电功能材料。
本发明的技术方案:
一类给受体型芴基纳米格子是以芴基衍生物和电子受体单元交替排列成方环形的刚性结构,其结构如通式(I)所示:
其中:
所述W为C或N;
所述Ar1、Ar2、Ar3、Ar4相同或不同,各自独立地代表共轭芳环单元;
所述G1、G2为芳香基团或者非芳香基团。
进一步地,所述Ar1、Ar2的结构相同或不同,分别选自如下单元中的一种:
其中,m代表重复单元个数,为2~100的自然数;
所述Ar3、Ar4的结构相同或不同,分别选自如下单元中的一种:
以上各式中所述R1~R8为氢、未取代或含有取代基的C1~12烷基、未取代或含有取代基的C1~12烷氧基;所述X为O、N、S或Se。
此处代表单元的连接位点。
给受体型芴基衍生物纳米格子分子的制备方法,通过酸催化的Friedel–Crafts反应一步成环制备,带有叔醇活性位点和芳香环端基活性位点的前体自身缩合,其反应路线如下:
具体反应步骤为:带有叔醇和芳香环端基氢双活性位点的前体溶解于有机溶剂中,在室温下加入催化剂搅拌反应,反应5min~12h,通过色谱柱分离得到产物。
进一步地,所述有机溶剂为二氯甲烷。
进一步地,所述催化剂为三氟化硼乙醚络合物。
本发明的给受体型芴基衍生物纳米格子分子可应用于有机/聚合物发光二极管,其中发光二极管器件的结构为透明阳极/发光层/电子注入层/阴极,发光层由主体材料与掺杂客体组成,纳米格子分子作为主体材料。
给受体型芴基衍生物纳米格子分子可应用于信息存储器件,其中存储器件的结构低栅顶接触,顺序可以为基底、栅极、遂穿层、有机半导体、源极和漏极,其中格子分子作为介电层,通过真空蒸镀、溶液旋涂或喷墨打印方式制备。
给受体型芴基衍生物纳米格子分子还可应用于有机忆阻器,其中电池器件结构为本体异质结型,忆阻器件的结构为典型的三明治结构,顺序可以为ITO、活性层、Al电极,半导体材料作为活性层,通过真空蒸镀、溶液旋涂或喷墨打印方式制备。
给受体型芴基衍生物纳米格子分子还可应用于有机太阳能电池,其中电池器件结构为本体异质结型,顺序可以为ITO阳极、PEDOT/PSS修饰层、活性层、Al阴极,其中格子分子和PCBM或者PC71BM共混旋涂作为活性层。
有益效果:
本发明通过核磁共振氢谱和碳谱(1H NMR、13C NMR)、飞行时间质谱(MALDI-TOF MS)等表征给受体型芴基衍生物纳米格子分子的结构,通过热重分析和差热分析测试了材料的热稳定性,通过循环伏安法表征了它们的电化学性质,通过薄膜高温退火的方式测试了材料的光谱稳定性。通过以上手段对给受体型芴基衍生物纳米格子分子的测试,其结果表明该类芴基格子分子表现出良好的热稳定性;光谱测试显示此类材料具有较小的斯托克斯位移,光谱的溶液依赖性小,分子间堆积紧密,薄膜退火光谱显示其具有优异的光谱稳定性;电化学测试显示此类材料是典型的p型材料,材料的HOMO能级低于空气的氧化门槛,具有良好的电化学稳定性。
本发明提供的纳米格子材料可以作为高效的发光主体材料、空穴传输或电子阻断材料或光伏活性层材料。该类芴基格子分子的可溶液加工、孔径的可精确调控性、能级、带隙的可调性的特点使得该类半导体材料适用于有机场效应晶体管存储器、有机发光二极管、有机电致发光、光伏电池、光探测、化学与生物传感和有机激光等领域的应用。本发明的主要优点在于:
(1)合成方式模块化,高拓展性;
(2)刚性框架结构提供高的热学、电化学、光学稳定性等优点;
(3)框架的刚性结构可减少器件制备过程中的薄膜溶剂依赖性;
(4)相比于COFs、MOFs分子,此类环状材料可以实现大面积可溶性加工;
(5)通过改变桥连分子片段可以精确调控孔径尺寸,孔径在1nm左右,具有纳米级尺寸;
(6)引入不同电子受体单元可以实现对带隙、能级排布的精确调控等优点;
(7)给受体的引入格子分子可以和受体材料进行相互作用,在有机光伏器件或者主客体掺杂器件中能够展现较好的性能。
附图说明
图1为纳米格子G1的核磁氢谱图;
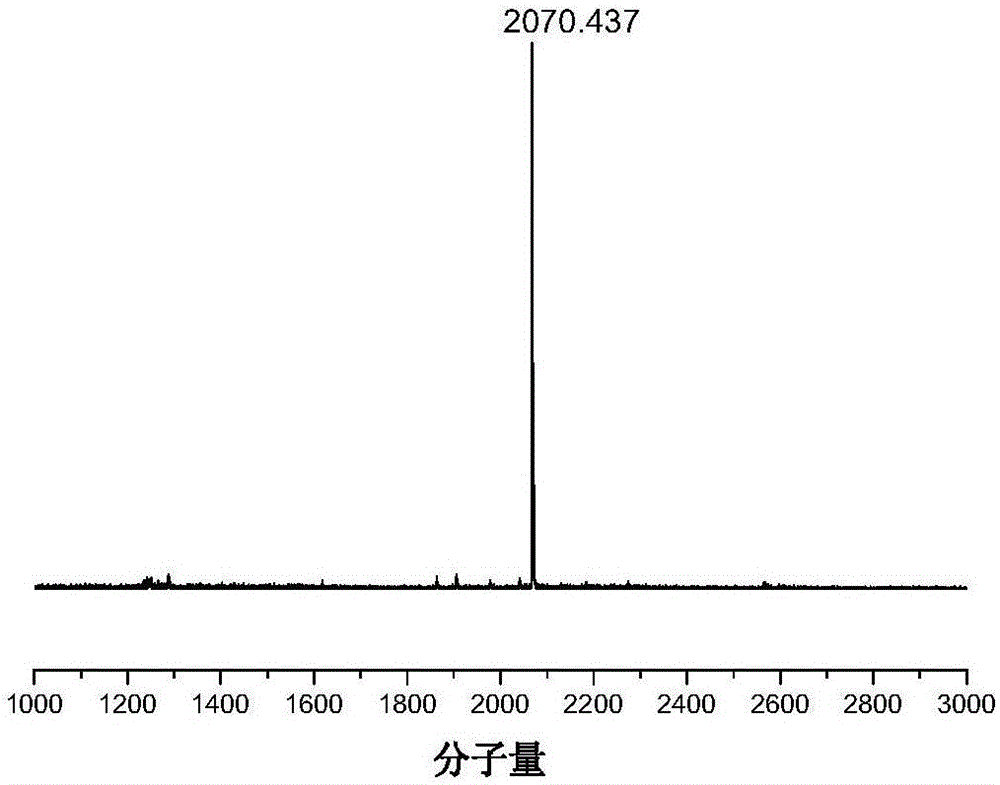
图2为纳米格子G1的飞行时间质谱图;
图3为纳米格子G2的核磁氢谱图;
图4为纳米格子G2的飞行时间质谱图;
图5为纳米格子G1在二氯甲烷中的紫外吸收光谱和荧光发射光谱;
图6为纳米格子G2在二氯甲烷中的紫外吸收和荧光发射光谱;
图7为纳米格子G1的循环伏安曲线;
图8为纳米格子G1正向扫描11V的忆阻性能曲线;
图9为纳米格子G1正向扫描10V的忆阻性能曲线。
具体实施方式
下面通过具体实施方式对本发明作进一步详细说明。但本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
本发明实施例提供的给受体型芴基纳米格子材料是通过酸催化的Friedel–Crafts反应一步成环制备得到,带有叔醇活性位点和芳香环端基活性位点的前体自身缩合,其反应路线如下:
其中:
所述W为C或N;
所述Ar1、Ar2、Ar3、Ar4相同或不同,各自独立地代表共轭芳环单元;
所述G1、G2为芳香基团或者非芳香基团。
进一步地,所述Ar1、Ar2的结构相同或不同,分别选自如下单元中的一种:
其中,m代表重复单元个数,为2~100的自然数;
所述Ar3、Ar4的结构相同或不同,分别选自如下单元中的一种:
以上各式中所述R1~R8为氢、未取代或含有取代基的C1~12烷基、未取代或含有取代基的C1~12烷氧基;所述X为O、N、S或Se。
实施例1
当W为C;Ar1、Ar2为对溴辛氧基苯,Ar3为二联噻吩或者三联噻吩,Ar4为苯并噻二唑,G1、G2均为氢,所述的格子分子结构分别如下:
反应路线如下:
具体制备方法为:二联噻吩、三联噻吩是由噻吩单体通过Pd/C催化下偶联得到,双溴代苯并二噻唑经高温硼酸化得到化合物3,单溴芴酮通过格式反应和Friedel–Crafts反应得到化合物5,化合物3、4、5在Pd(PPh3)4催化剂,碱溶液选择K2CO3/KF条件下Suzuki偶联,高效的得到单取代的L形前体。L形前体通过自身的双活性位点叔醇基团及噻吩的2位在Et2O·BF3催化下Friedel–Crafts反应闭环得到给受体型芴基格子G1或者G2。
化合物1:2-溴噻吩(5.0g,30.6mmol)、10%的Pd/C(1.6g,1.5mmol)加入80mL无水THF,氮气保护,溶液升温至80℃,回流24小时。溶液冷却至室温,倒入80mL去离子水,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚),得到白色固体产物1(4.4g,87%)。GC-MS(EI-m/z):166.3/166(M+).1H NMR(400MHz,CDCl3,ppm):δ7.25(m,4H),7.08-7.06(m,2H).
化合物2:镁条(1.49g,62.1mmol)加入20mL的无水THF,氮气保护,加入几粒碘,将溶解有2-溴噻吩(8.4g,51.8mmol)的30mL无水THF逐滴加入上述溶液,用电吹风局部加入反应,使溶液持续平稳的回流1小时,冷却至室温,得到相应的格式试剂。将2,5-二溴噻吩(10.4g,43.1mmol)、Ni(dppp)Cl2(0.46g,0.58mmol)加入50mL无水的THF中,把之前制备的格式试剂用注射器转移到上述溶液中,溶液升温至70℃搅拌过夜。溶液冷却至室温,倒入50mL稀盐酸中,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚),得到白色固体2(7.7g,72%)。GC-MS(EI-m/z):248.4/248(M+),1H NMR(400MHz,CDCl3)δ(ppm):7.76(d,J=2.0Hz,2H),6.98(d,J=2.0Hz,2H).
化合物3:联硼酸频那醇酯(10.5g,41mmol)、4,7-二溴-2,1,3-苯并噻二唑(5g,17mmol)、KOAc(3.3g,34mmol)、Pd(dppf)Cl2(0.63g,8.6mmol)加入到带有冷凝管的100ml烧瓶中,抽真空通氮气三次,注射器注入1,4-二氧六环100ml,回流反应18h。反应完毕后,二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚:乙酸乙酯混合溶剂,v/v=4:1),得到略带淡黄色的固体3(5.90g,88.6%)。GC-MS(EI-m/z):388.1/388(M+)。1H NMR(400MHz,CDCl3,ppm):δ8.13(s,2H),1.44(s,24H).
化合物4:镁条(0.6g,25.5mmol)加入20mL的无水THF,氮气保护,加入几粒碘,将溶解有1-溴-4-(辛氧基)苯(6.1g,21.2mmol)的20mL无水THF逐滴加入上述溶液,用电吹风局部加入反应,使溶液持续平稳的回流1小时,冷却至室温,得到相应的格式试剂。将2,7-二溴-9H-芴-9-酮(6.5g,19.3mmol)加入50mL无水的THF中,把之前制备的格式试剂用注射器转移到上述溶液中,溶液升温至70℃搅拌过夜。溶液冷却至室温,倒入50mL饱和氯化铵溶液中,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚:二氯甲烷(1:1,v/v),得到目标产物4(8.6g,82%)。GC-MS(EI-m/z):464/466(M+).1H NMR(400MHz,CDCl3,ppm):δ7.63(d,J=7.6Hz,1H),7.48(m,3H),732(m,5H),6.80(d,J=8.8Hz,2H),3.90(t,J=7.6Hz,2H),2.51(s,1H),1.75(m,2H),1.42(m,2H),1.27(m,8H),0.87(t,J=7.6Hz,3H).
化合物5a和5b:250ml双口烧瓶加入4(0.5g,0.92mmol)的二氯甲烷溶液(150ml),将化合物1(0.12g,0.74mmol)和催化剂BF3.Et2O(1.3g,9.2mmol)的二氯甲烷(50mL)溶液置于恒压滴液漏斗中逐滴加入上述溶液。滴加完毕后,待TLC板监控反应完毕,倒入80mL去离子水,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚/二氯甲烷,v/v=8:1),得到淡黄色油状产物5a(0.36g,80%)。1H NMR(400MHz,CDCl3)δ(ppm):7.73(d,J=7.2Hz,1H),7.62(m,2H),7.49(dd,J=8.0Hz,2.0Hz,2H),7.34(m,2H),7.14(m,3H),7.05(dd,J=4.5Hz,0.8Hz,1H),6.94(m,2H),6.77(d,J=8.8Hz,2H),6.72(d,J=4.0Hz,1H),3.91(t,J=6.4Hz,2H),1.75(m,2H),1.42(m,2H),1.30(br,8H),0.88(m,3H).
合成过程类似于5a,可以得到产黄色物油状产物5b(80%)。MALDI-TOF-MS(m/z):695.8/696[M+].1H NMR(400MHz,CDCl3)δ7.79–7.70(m,1H),7.62(dd,J=9.1,3.6Hz,2H),7.55–7.46(m,2H),7.43–7.28(m,2H),7.22–7.10(m,4H),7.04–6.97(m,2H),6.97–6.86(m,2H),6.79–6.70(m,3H),3.96–3.85(m,2H),1.81–1.10(m,15H).
化合物6a和6b:化合物4(0.3g,0.64mmol)、化合物3(0.25g,0.64mmol)、化合物5a(0.39g,0.64mmol)、Pd(PPh3)4(37.0mg,0.032mmol)加入150ml圆底烧瓶,K2CO3(10ml,2M)和甲苯(15ml)在使用前分别通入氮气30min以除去溶液中的氧气。将上述溶液分别注入圆底烧瓶中,溶液升温至100℃,回流48小时。溶液冷却至室温,倒入80mL去离子水,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚/二氯甲烷,v/v=2:1),得到淡黄色粉末固体6a(0.23g,35%)。MALDI-TOF-MS(m/z):1052.4/1053.1[M+].1H NMR(400MHz,CDCl3)δ(ppm):8.05(m,3H),7.92(m,2H),7.82(t,J=7.6Hz,2H),7.72(m,3H),7.58(d,J=7.2Hz,2H),7.41(m,6H),7.29(m,3H),7.14(dd,J=5.2Hz,1.2Hz,1H),7.05(dd,J=3.6Hz,0.8Hz,1H),6.94(m,2H),6.85(d,J=4.0Hz,1H),6.78(t,J=8.4Hz,4H),3.88(t,J=6.4Hz,4H),2.68(s,1H),1.74(m,4H),1.30(br,20H),0.88(t,J=6.4Hz,6H).
合成过程类似于6a,可以得到产黄色固体6b(30%)。MALDI-TOF-MS(m/z):1135.6/1135.427[M+].1H NMR(400MHz,CDCl3)δ8.09(m,3H),7.92(m,2H),7.82(m,2H),7.70(m,3H),7.58(d,J=7.6Hz,1H),7.43(m,6H),7.28(m,3H),7.17(dd,J=4.8Hz,0.8Hz,1H),7.12(dd,J=3.6Hz,1.2Hz,1H),6.95(m,4H),6.87(d,J=4.0Hz,1H),6.78(m,4H),3.88(t,J=6.4Hz,4H),2.79(s,1H),1.72(s,4H),1.39(br,20H),0.88(m,6H).
纳米格子G1和纳米格子G2:250ml双口烧瓶加入化合物6a(0.2g,0.19mmol)的二氯甲烷溶液(150ml),将BF3.Et2O(0.14g,0.95mmol)的二氯甲烷(50mL)溶液置于恒压滴液漏斗中逐滴加入上述溶液。滴加完毕后,待TLC板监控反应完毕,倒入80mL去离子水,用二氯甲烷萃取,合并有机层用水和饱和食盐水洗涤,之后用无水硫酸镁干燥,过滤,用旋蒸仪除去溶剂,将粗产物用层析柱色谱分离(洗脱剂为石油醚/二氯甲烷,v/v=4:3),得到黄色固体产物(20mg,10%)。MALDI-TOF-MS(m/z):2070.9/2070.437[M+].1H NMR(400MHz,CDCl3)δ8.17(s,4H),7.95(d,J=8.0Hz,4H),7.88(d,J=8.0Hz,4H),7.88(d,J=7.6Hz,4H),7.71(s,4H),7.50(d,J=7.2Hz,4H),7.40(t,J=7.2Hz,4H),7.32(t,J=7.2Hz,4H),7.25(d,J=8.8Hz,8H),6.81(d,J=3.6Hz,4H),6.75(d,J=8.8Hz,8H),6.64(d,J=3.6Hz,4H),3.83(m,8H),1.64(m,8H),1.20(m,40H),0.88(m,12H).
合成过程类似于纳米格子G1,可以得到黄色固体产物纳米格子G2(10%)。MALDI-TOF-MS(m/z):2235.1/2235.965[M+].1H NMR(400MHz,CDCl3)δ8.19(m,2H),8.06(m,5H),7.97(m,6H),7.83(m,9H),7.54(m,5H),7.40(m,4H),7.31(m,5H),7.26(m,4H),6.82(m,20H),3.85(m,8H),1.71(m,8H),1.34(br,40H),0.85(m,12H).
实施例2
热重分析(TGA)在岛津公司(Shimadzu)DTG-60H热重分析仪上进行,加热扫描速度为10℃/min并且氮气流速为20cm3/min。示差扫描量热分析(DSC)在岛津公司(Shimadzu)DSC-60A测试仪上进行,样品首先以10℃/min的速度加热到样品分解温度低十度的状态,然后,在液氮条件下降温回到开始温度,同样第二次以10℃/min的速度加热升温扫描。从TGA实验中可得失重5%时的温度(Td)分别为355℃,320℃。DSC实验显示无明显玻璃化转变温度。
实施例3
把纳米格子G1、G2分别配成准确的1μM的二氯甲烷稀溶液,并通过氩气冲洗去掉氧气。采用岛津UV-3150紫外可见光谱仪和RF-530XPC荧光光谱仪进行吸收光谱和发射光谱测定。
如图5、6所示,纳米格子G1在二氯甲烷中紫外吸收光谱和荧光光谱的峰值分别为381nm,530nm。纳米格子G2在二氯甲烷中紫外吸收光谱和荧光光谱的峰值分别为408nm,534nm。
实施例4
对纳米格子G1、G2材料的电化学测定,在上海辰华仪器公司的CHI 660D型电化学工作站测得,采用三电极体系Pt片电极作为工作电极,铂丝为辅助电极,甘汞电极为参比电极。测试时以0.1M四丁基氟硼酸胺(Bu4NPF6)的乙腈溶液为电解质溶液,二茂铁作为内标(0.40V),电解质溶液使用前通入氮气20分钟除去氧气,将聚合物的氯仿溶液滴涂在工作Pt电极上成膜,测试时扫描速度为50mV/s。根据扫描的过程中会产生氧化峰和还原峰,通过分析出现的氧化峰或还原峰的起始氧化/还原电位,进而计算出HOMO能级和LUMO能级。
如图7所示,电化学测试显示纳米格子G1的HOMO、LUMO能级分别为-5.97eV,-4.0eV。纳米格子G2的HOMO、LUMO能级分别为-5.98eV,-4.2eV。
实施例5
本发明提供了一种三明治结构的有机忆阻器件,器件结构为ITO/纳米格子G1掺杂离子盐(50nm)/Al。该器件包括:ITO阳极、活性层、Al阴极。获得了忆阻性能良好的有机二极管器件。如图8~9所示,二极管器件实现了不同扫描电压的学习规律曲线,正向扫描电压为10V条件下,器件扫描曲线逐渐减小,实现了“学习规律”中的遗忘性能;正向扫描电压为11V条件下,器件扫描曲线逐渐增加,实现了“学习规律”中的学习性能。
总之,以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所作的均等变化与修饰,皆应属本发明专利的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!