一种富马酸沃诺拉赞新晶型及其制备方法与流程
本发明涉及医药技术领域,具体涉及一种富马酸沃诺拉赞新晶型及其制备方法。
背景技术:
富马酸沃诺拉赞(Vonoprazan fumarate,TAK-438),化学名为1-[5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1H-吡咯-3-基]-N-甲基甲胺单富马酸盐,是日本武田制药公司研发的一种新型钾离子通道阻滞剂,于2015年在日本上市,用于治疗非糜烂性胃食管反流病、十二指肠溃疡、胃溃疡、糜烂性食管炎等。化学结构式如(1)所示。
富马酸沃诺拉赞以浓度依赖性的方式抑制基础胃酸的分泌,ID50值为1.26mg/kg。在与兰索拉挫对比的动物体内实验中,富马酸沃诺拉赞抑制胃酸分泌的作用更强,升高胃分泌物pH值的速度更快,有效浓度可持续至给药后5小时以上。当剂量为1mg/kg时,pH值在给药后90分钟时达到最高峰,达到的最高pH值为5.9。
经检索,文献(CN101300229A,实施例47;WO026916A1,实施例69;Journal of Medicinal Chemtry,2012,55,pp4446-4456))等报道的富马酸沃诺拉赞合成工艺中,5-(2-氟苯基)-1H-吡咯-3-甲醛为关键的中间体,其经磺酰化、和甲胺缩合形成亚胺、最后还原得沃诺拉赞游离碱、与富马酸成盐后经甲醇-水精制得富马酸沃诺拉赞,反应合成路线如(2)所示。
CN105315258A采用乙酸乙酯/甲醇或醇/水精制获得两种晶型,但富马酸沃诺拉赞药学评审报告中提到未发现多晶型现象。
富马酸沃诺拉赞属于难溶性药物,在制剂中一般以固体形式应用,且其对光照较敏感,因此对其晶型的研究具有十分重要的意义。另外,药物中存在的杂质对产品的质量影响较大,因而恰当的除杂方法十分必要。
技术实现要素:
本发明的目的在于提供一种富马酸沃诺拉赞新晶型及其除杂方法,该晶型的粉末X-衍射图中,在衍射角2θ位于12.18±0.5、13.43±0.5、22.35±0.5、37.06±0.5、38.25±0.5处有特征峰。
进一步地,所述晶型的粉末X-衍射图中,还在衍射角2θ位于15.02±0.5、15.22±0.5、20.33±0.5处有特征峰。
更进一步地,所述晶型的粉末X-衍射图中,各特征峰的峰强度为:
其中,所述晶型的DSC吸热峰在210±5℃,优选为210±2℃。
本发明还提供了所述新晶型的制备方法,包括如下步骤:
(1)取富马酸沃诺拉赞粗品,加入至乙酸乙酯、甲醇和纯化水的混合溶剂中,加热使固体溶解;其中所述富马酸沃诺拉赞粗品和混合溶剂的重量体积比为1:5~20g/ml,更优选10~15g/ml;
其中所述混合溶剂为乙酸乙酯、甲醇和纯化水,其体积比为1:0.1~1:0.01~0.1(V/V),更优选1:0.3~0.5:0.05~0.08(V/V)。
优选的,所述富马酸沃诺拉赞粗品的溶解温度为50~100℃,更优选60~70℃。
(2)过滤,滤液降温析晶,过滤、干燥得白色结晶。
优选的,析晶温度为0~20℃,更优选0~5℃。
优选的,干燥温度为40~70℃,更优选50~60℃。
本发明的有益效果是:
本发明提供了富马酸沃诺拉赞的制备方法,能显著降低富马酸沃诺拉赞粗品中工艺副产物的含量,采用新的精制工艺就可以获得达到质量要求的高纯度富马酸沃诺拉赞,HPLC检测富马酸沃诺拉赞纯度达到99.9%以上,单一杂质含量均小于0.05%。
本发明的富马酸沃诺拉赞新晶型制备中,日本武田评审报告中提到的合成副反应杂质U2\U3\U4均检测不出,只存在U6\U9 2个杂质,含量在0.1%以下。
合成副反应杂质的结构式为:(1)杂质U2;(2)杂质U3;(3)杂质U4;(4)杂质U6;(5)杂质U9。
本发明制备的富马酸沃诺拉赞新晶型,可显著降低对光、热的不稳定,避免了因光照、高温高湿产生的降解,为该药物的储存、运输提供了便利,为提高该药物的稳定性及研究其溶出度提供了基础。
附图说明
附图1为富马酸沃诺拉赞新晶型X-衍射图谱;
附图2为富马酸沃诺拉赞新晶型DSC图谱;
附图3为富马酸沃诺拉赞对照晶型X-衍射图谱;
附图4为富马酸沃诺拉赞新晶型HPLC图谱;
附图5为富马酸沃诺拉赞对照晶型HPLC图谱。
具体实施方式
以下结合具体实施例和附图详细说明本发明,目的是使本发明的优点更翔实,而非限制本发明。本领域技术人员应该理解,本发明并不限于这些实施例以及使用的制备方法,任何对本发明进行同等替换、组合、改良或修饰,均包含于本发明内。
本发明中使用的富马酸沃诺拉赞粗品,可购自于市售产品,也可通过现有技术合成得到。
实施例
富马酸沃诺拉赞新晶型及对照晶型的制备
(1)富马酸沃诺拉赞中间体的合成
取5-(2-氟苯基)-1H-吡咯-3-甲醛300g,4-二甲氨基吡啶37.5g,N,N-二异丙基乙胺300g,乙腈1L,加入反应瓶中。将吡啶-3-磺酰氯300g溶于乙腈1L中,并在20~30℃下滴加到上述反应体系中,约1小时滴完。滴完后体系升温至40~50℃,继续1小时反应至原料消耗完全。体系降温至30℃,滴加纯化水1.8L淬灭反应,用0.5mol/L HCl调节pH至4~5,析出黄色固体。向体系加入纯化水3.6L,于0~10℃搅拌1小时。抽滤,滤饼用乙腈:纯化水(1:2)900ml及纯化水900ml洗涤,50℃减压烘干得棕色粉末固体475g,收率90.6%。
(2)富马酸沃诺拉赞粗品的合成
将5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1H-吡咯-3-甲醛330g、甲醇1.6L加入反应瓶中,搅拌使固体溶解。控温在20℃以下滴加含有30%甲胺的甲醇溶液145ml,滴毕继续搅拌30分钟。分批加入硼氢化钠18.6g,在室温下搅拌2小时,后处理得油状物,加入富马酸114g成盐得富马酸沃诺拉赞粗品312g,收率为67.6%。
(3)富马酸沃诺拉赞新晶型的制备
将上述粗品160g加入乙酸乙酯1.6L、甲醇0.64L和纯化水0.1L中,加热至65℃使固体溶解,过滤,冷却至0~5℃析晶6小时,抽滤,50℃减压干燥得白色结晶121.9g,收率76.2%,单一杂质0.027%,总杂质0.043%。
(4)对照晶型的制备
将上述粗品150g加入甲醇1L和纯化水0.5L中,加热至60℃使固体溶解,过滤,冷却至0~5℃析晶7小时,抽滤,50℃减压干燥得白色结晶108.3g,收率为72.2%,单一杂质0.061%,总杂质0.12%。
富马酸沃诺拉赞新晶型及对照晶型的检测
一、富马酸沃诺拉赞新晶型和对照晶型粉末X-衍射测定
粉末X-衍射测定仪器参数:靶型:CuKɑ线;(单色器);管电压40KV;管电流30mA;狭缝:1.0/1.0/Ni/0.2;步长:0.02°;扫描范围(2θ):3.00~40.00°;扫描速度:10.00Deg.min-1。
所得新晶型的粉末X-衍射测定数据见表1:
表1
所得对照晶型的粉末X-衍射测定数据见表2:
表2
其中测定结果见附图1、图3。
二、富马酸沃诺拉赞新晶型DSC检测
取富马酸沃诺拉赞新晶型,进行DSC检测,测定结果见附图2。
三、新晶型及对照晶型的HPLC检测
检查方法:中国药典2015版二部附录高效液相色谱法
试验条件:C18柱(型号:长150mm,内径4.6mm,填充料粒径3μm);检测器:UV检测器;检测波长:230nm;柱温:25℃;流速:1.0ml/min;流动相A:0.025mol/L磷酸钠溶液(用磷酸调节pH值至7.2)、流动相B:乙腈、流动相C:甲醇,线性梯度洗脱,具体洗脱程序如表3:
表3
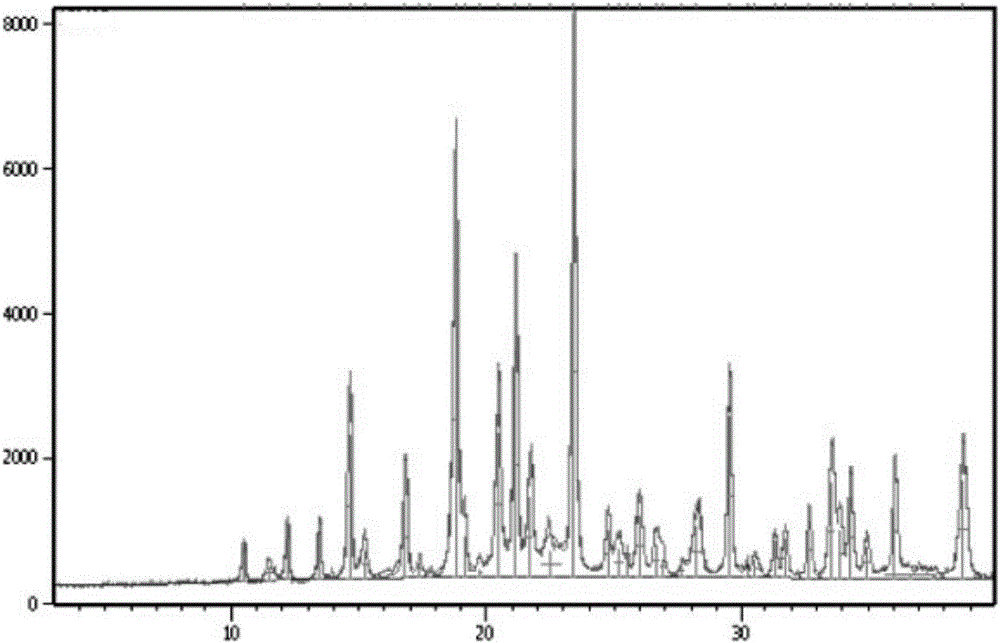
具体操作:精密量取供试品溶液20μl,注入液相色谱仪,记录色谱图。供试品溶液色谱图中如有杂质峰,按峰面积归一化法计算,新晶型检测数据见表4,对照晶型见表5,HPLC图谱见附图4、图5:
表4
其中,保留时间1.600min为富马酸,保留时间18.769min为U6,保留时间36.972min为U9。
表5
以下通过影响因素试验说明本发明的有益效果:
将实施例1制备得的本发明的晶型与对照晶型(参考CN101300229A)分别进行影响因素试验,分别在25℃/RH60%长期条件下6个月,40℃/RH75%加速条件下6个月,其结果分别见表6,7:
表6
表7
从上述HPLC和影响因素的结果可知,采用本发明所制备的富马酸沃诺拉赞新晶型,比对照晶型相对更稳定,其制备方法能更显著降低富马酸沃诺拉赞粗品中工艺杂质的含量。
- 还没有人留言评论。精彩留言会获得点赞!