富马酸‑CO‑释放分子杂合物、其在治疗炎性或心血管疾病中的用途和其制备方法与流程
本发明涉及能够活化血红素加氧酶-1(HO-1)活性和HO-1蛋白质表达的杂合富马酸-CO-释放分子、其合成和其在治疗应用中的用途,特别是其在治疗炎性或心血管疾病中的用途。
背景技术:
HO-1/CO系统是防御体系的重要部分,所述防御体系对抗由重金属、活性氧物质、脂多糖(LPS)和其他炎性过程介导的各种应激情况造成的损伤,因此所述系统在细胞保护和抗炎反应的调节中发挥关键作用。
在心血管功能障碍、肺动脉高压和炎性病症如败血症和炎症性肠病的模型中,已显示以低剂量(250ppm)持续受限制的一段时间施用CO气体使HO-1的许多有益作用重现(恢复)。
由于对HO-1/CO治疗潜力的兴趣,向生物系统放出受控制量的CO(CO-释放分子,CO-RMs)的化合物已被开发,并且被证明发挥与CO释放有关的广泛的药理作用。文献中描述的绝大多数有药理活性的CO-RMs为含有Ru、Fe、Mn、Co和Mo之一的金属羰基化合物。
转录因子Nrf2为细胞应激反应的至关重要的引发剂,因为其协调一些抗氧化剂和解毒基因的表达,所述抗氧化剂和解毒基因修复损伤并恢复细胞稳态。作为这种可诱导的反应的一部分,血红素加氧酶-1(HO-1)通过利用血红素产生CO、胆绿素/胆红素和铁、对抗氧化应激和炎症的重要信号和保护分子发挥突出的作用。
已确认许多化学物质发挥Nrf2/HO-1活化剂的作用,并且在它们当中,一些具有α,β-不饱和羰基官能团的天然衍生的化合物,包括姜黄素(发现增加HO-1表达的第一种化学物质)和查耳酮。Nrf2/HO-1活化剂的名单现在已经增长到包括数百个化合物。由于它们的作用机制,Nrf2/HO-1活化剂需要时间以发动细胞应激反应,导致延迟的、尽管必要的有益作用。
在疾病中,相比于单独的CO-RMs或Nrf2活化剂,具有快速释放CO并同时刺激更持久的Nrf2依赖性保护蛋白质组的能力的分子的合成提供了显著的治疗优势,在所述疾病中这些通路对于组织保护是至关重要的,因此有对这种分子的需要。
Zhu等人(J.Organomet.Chem.2006,691,485-490)公开了化合物[Co2(CO6)(μH2CCH2O)-]2(CO)2,然而仅涉及到其在含有C2Co2R2的混合的金属连接的炔烃桥接的蝶形原子簇(butterfly cluster)中的用途,以及在聚合材料的合成中的应用。
WO 2012/076696公开了结合至CO-释放分子的姜黄素衍生物。然而,本发明的发明人已经表明这些分子不释放一氧化碳。这种CO-释放的缺乏导致分子不显示出与通过CO活化HO-1有关的广泛的有益的治疗作用。
WO 2008/003953还描述了式(I)的CO-释放分子:Mn(CO)4XY用于向人和动物治疗性递送CO,其中X和Y为单齿配体,或者一起形成二齿配体。更具体地,WO 2008/003953公开了CO-释放化合物364,其中X代表O(O)CCH2CH2C(O)OH并且Y代表Br。然而,该分子被证明具有不良的CO-释放能力。
技术实现要素:
出乎意料地,已发现包含富马酸部分和CO-释放分子(CORM)的杂合分子(hybrid molecule)能够快速放出CO并活化Nrf2/HO-1通路,导致细胞中炎性防御体系的双重活化和改进的治疗效果。
本发明涉及具有一个CORM部分的化合物(单-CORM化合物)和具有两个CORM部分的化合物(二-CORM化合物)。本发明人已发现两种类型的分子都具有改进的治疗活性。此外,已观察到单-CORM分子非常快地释放高水平的CO,因此发挥“突释效应”。另一方面,二-CORM化合物释放更高水平的CO(特别地是由于第二个CORM部分的存在),但不那么快且持续较长的一段时间。因此,当寻求“突释效应”时,将使用本发明的单-CORM化合物,而当持续很长一段时间的释放对于患者或待治疗的疾病或病症更为合适时,将使用本发明的二-CORM化合物。
因此,本发明涉及式(I)的杂合富马酸-一氧化碳释放分子(富马酸-CORM)、其药学上可接受的盐、水合物和溶剂化物:
其中:
A代表:
·单键,或者
·-Z-Q-,其中:
οQ代表O,S或NR2,其中R2代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基或(C1-C6)烷基-(C3-C8)杂环基,-(C3-C14)环烷基,或R2和Z相连以形成(C3-C8)杂环基,
οZ代表-(C1-C6)烷基-,-(C2-C6)烯基-,-芳基-,-杂芳基-,-(C2-C6)炔基-,-(C3-C8)杂环基-,-(C3-C14)环烷基,-(C1-C6)烷基-R3-(C1-C6)烷基-,-(C2-C6)烯基-R3-(C1-C6)烷基-,-(C1-C6)烷基-R3-(C2-C6)烯基-,-(C2-C6)烯基-R3-(C2-C6)烯基-,-(C1-C6)烷基-R3-(C2-C6)炔基-,-(C2-C6)烯基-R3-(C2-C6)炔基-,-(C2-C6)炔基-R3-(C2-C6)炔基-,-(C2-C6)炔基-R3-(C1-C6)烷基-,-(C2-C6)炔基-R3-(C2-C6)烯基-,-(C1-C6)烷基-OCO-,或-CH2-(CHOR3)CH2O-(C1-C6)烷基-,其中R3代表芳基,杂芳基,(C3-C8)杂环基,或(C3-C14)环烷基,
CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Co,Ru,Mn,Mo,V或Fe,优选Co,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价,
优选地,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
以及R1代表:
·-Q’-Y,其中
οQ’代表O,S或NR5,其中R5代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基或(C1-C6)烷基-(C3-C8)杂环基,-(C3-C14)环烷基,
οY代表H,-(C1-C6)烷基,-(C2-C6)烯基,-芳基,-杂芳基,-(C2-C6)炔基,-(C3-C8)杂环基,-(C3-C14)环烷基,-(C1-C6)烷基-R6-(C1-C6)烷基,-(C2-C6)烯基-R6-(C1-C6)烷基,-(C1-C6)烷基-R6-(C2-C6)烯基,-(C2-C6)烯基-R6-(C2-C6)烯基,-(C1-C6)烷基-R6-(C2-C6)炔基,-(C2-C6)烯基-R7-(C2-C6)炔基,-(C2-C6)炔基-R6-(C2-C6)炔基,-(C2-C6)炔基-R6-(C1-C6)烷基,-(C2-C6)炔基-R6-(C2-C6)烯基,(C1-C6)烷基-R6,-(C2-C6)烯基-R6,-(C2-C6)炔基-R6,其中R6代表芳基,杂芳基,(C3-C8)杂环基,或(C3-C14)环烷基,-CH2(CHOR6)CH2-OR7,其中R7代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基,(C1-C6)烷基-(C3-C8)杂环基,(C1-C6)烷基-(C3-C14)环烷基,
或者,
·A’-CORM’其中A’和CORM’分别如A和CORM所定义。
有利地,CORM选自:
Mn(CO)5,
更有利地选自:
甚至更有利地选自
优选地,本发明涉及以上式(I)的杂合富马酸-一氧化碳释放分子(富马酸-CORM)、其药学上可接受的盐、水合物和溶剂化物,其中:
A代表:
·单键,或者
·-Z-Q-,其中:
οQ代表O,S或NR2,其中R2代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基或(C1-C6)烷基-(C3-C8)杂环基,-(C3-C14)环烷基,或R2和Z相连以形成(C3-C8)杂环基,
οZ代表-(C1-C6)烷基-,-(C2-C6)烯基-,-芳基-,-杂芳基-,-(C2-C6)炔基-,-(C3-C8)杂环基-,-(C3-C14)环烷基,-(C1-C6)烷基-R3-(C1-C6)烷基-,-(C2-C6)烯基-R3-(C1-C6)烷基-,-(C1-C6)烷基-R3-(C2-C6)烯基-,-(C2-C6)烯基-R3-(C2-C6)烯基-,-(C1-C6)烷基-R3-(C2-C6)炔基-,-(C2-C6)烯基-R3-(C2-C6)炔基-,-(C2-C6)炔基-R3-(C2-C6)炔基-,-(C2-C6)炔基-R3-(C1-C6)烷基-,-(C2-C6)炔基-R3-(C2-C6)烯基-,-(C1-C6)烷基-OCO-,或-CH2-(CHOR3)CH2O-(C1-C6)烷基-,其中R3代表芳基,杂芳基,(C3-C8)杂环基,或(C3-C14)环烷基,
R1代表:
·-Q’-Y,其中
οQ’代表O,S或NR5,其中R5代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基或(C1-C6)烷基-(C3-C8)杂环基,-(C3-C14)环烷基,
οY代表H,-(C1-C6)烷基,-(C2-C6)烯基,-芳基,-杂芳基,-(C2-C6)炔基,-(C3-C8)杂环基,-(C3-C14)环烷基,-(C1-C6)烷基-R6-(C1-C6)烷基,-(C2-C6)烯基-R6-(C1-C6)烷基,-(C1-C6)烷基-R6-(C2-C6)烯基,-(C2-C6)烯基-R6-(C2-C6)烯基,-(C1-C6)烷基-R6-(C2-C6)炔基,-(C2-C6)烯基-R7-(C2-C6)炔基,-(C2-C6)炔基-R6-(C2-C6)炔基,-(C2-C6)炔基-R6-(C1-C6)烷基,-(C2-C6)炔基-R6-(C2-C6)烯基,(C1-C6)烷基-R6,-(C2-C6)烯基-R6,-(C2-C6)炔基-R6,其中R6代表芳基,杂芳基,(C3-C8)杂环基,或(C3-C14)环烷基,-CH2(CHOR6)CH2-OR7,其中R7代表H,(C1-C6)烷基,芳基,杂芳基,(C1-C6)烷基-芳基,(C1-C6)烷基-杂芳基,(C1-C6)烷基-(C3-C8)杂环基,(C1-C6)烷基-(C3-C14)环烷基,
或者,
·A’-CORM’其中A’和CORM’分别如A和CORM所定义,
以及,当R1代表-Q’-Y时,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Co,Ru,Mn,Mo,V或Fe,优选Co,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价;
以及,当R1代表A’-CORM’时,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Co,Ru,Mn,Mo,V或Fe,优选Fe,Co,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价;
优选地,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Ru,Mn,Mo,V或Fe,优选Fe,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价;
甚至优选地,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Fe或Mn,以及
n为选定的整数,所述整数使金属M没有自由价。
甚至更优选地,本发明涉及式(Ia)的杂合富马酸-一氧化碳释放分子(富马酸-CORM)、其药学上可接受的盐、水合物和溶剂化物:
其中A、CORM和-Q’-Y如式(I)中所定义。
在CORM代表下式的羰基金属络合物的情况下:
M代表Rh,Co,Ru,Mn,Mo,V或Fe,优选Co,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价,更优选地
n为1至4的整数,所选定的整数使金属M没有自由价。特别地,当M代表Rh时,n代表2;当M代表Co时,n代表2;当M代表Ru时,n代表2;当M代表Mn时,n代表3;当M代表Mo时,n代表3;当M代表V时,n代表4;当M代表Fe时,n代表3。
有利地,Q代表O或NR2,优选代表O,并且R2代表H或(C1-C6)烷基。
有利地,Z代表-(C1-C6)烷基-,-(C2-C6)烯基-或-(C3-C8)杂环基-,-(C1-C6)烷基-R3-(C1-C6)烷基-,-(C2-C6)烯基-R3-(C1-C6)烷基-,并且R3代表杂芳基或(C3-C8)杂环基。
有利地,Q代表O,S或NR2,其中R2代表H,(C1-C6)烷基,并且Z代表-(C1-C6)烷基-,-(C2-C6)烯基-,-(C3-C8)杂环基-,-(C1-C6)烷基-R3-(C1-C6)烷基-,-(C2-C6)烯基-R3-(C1-C6)烷基-,其中R3代表杂芳基或(C3-C8)杂环基。
更有利地,Z代表:-CH2-CH2-,
有利地,CORM选自:
Mn(CO)5,
更有利地选自
Mn(CO)5,
甚至更有利地选自
有利地,Q’为O或NR5,优选为O。
有利地,Y选自H;-(C1-C6)烷基,-(C2-C6)烯基;-芳基,-杂芳基,-(C2-C6)炔基,-(C3-C8)杂环基;-(C3-C8)环烷基或-(C1-C6)烷基-芳基。
根据第一实施方案,本发明涉及式(Ia)的杂合富马酸-一氧化碳释放分子(富马酸-CO-RM),其中富马酸部分在一侧被CO-释放羰基络合物取代,并且在另一侧被酸、酯、硫酯或酰胺取代:
其中A,CORM和-Q’-Y如式(I)中所定义。
出乎意料地,已发现式(Ia)的化合物能够以很快的速度释放CO。例如,作为式(Ia)的化合物的例证,下面实施例中的化合物A能够在实施例中描述的体外条件下,以约38分钟的半衰期释放CO,从本发明的意义上说这是很快的速度。因此,式(Ia)的化合物对于相对快的生物活性是有利的。此外,实验证明仅包含一个CORM基团的本发明的化合物是有效的Nrf2的活化剂和HO-1蛋白质表达的诱导剂,并且其比富马酸二甲酯更有效。
根据该第一实施方案的一些有利的化合物具有式(Ia1):
其中:
M代表Mn(CO)4或Ru(CO)3Cl,以及
Q,Q’,W,Z和Y如式(I)中所定义。
有利地,在式(Ia1)的化合物中,Z-W为-CH2-CH2-NR7-,其中R7代表(C1-C3)烷基。
有利地,Q为O或NR2,优选为O。
有利地,Q’为O或NR5,优选为O。
有利地,Y代表H,-(C1-C6)烷基或-(C2-C6)烯基。
根据该第一实施方案的另一些有利的化合物具有式(Ia2):
其中:
Q’和Y如式(I)中所定义。
有利地,Y为H,-(C1-C6)烷基或-(C2-C6)烯基,并且Q’代表O。
根据该第一实施方案的一些进一步有利的化合物具有式(Ia3):
其中:
Q,Q’,Y和Z如式(I)中所定义。
有利地,Y为H,-(C1-C6)烷基或-(C2-C6)烯基,Q’为O,并且Q和Z如式(I)中所定义。更有利地,Q-Z为氨基酸(如丝氨酸,半胱氨酸,酪氨酸或赖氨酸)的侧链。
根据该第一实施方案的进一步有利的化合物具有式(Ia4):
其中:
Mx(CO)y代表Co2(CO)6或Co4(CO)10,
Q’,Q,Y和Z如式(I)中所定义。
有利地,Y为H,-(C1-C6)烷基或-(C2-C6)烯基,Q’为O,并且Q和Z如式(I)中所定义。
更有利地,Q为O,并且Z为-(C1-C6)烷基,优选为(C1-C3)烷基。
在第二实施方案中,本发明涉及杂合富马酸-一氧化碳释放分子(富马酸-CO-RM),其中富马酸部分在两侧均被CO-释放羰基络合物取代。
相比于单金属化合物,已发现这些双金属化合物持续很长一段时间释放较高量的CO,因此对于持久的生物活性是有利的。例如,作为以后描述的式(Ic)的化合物的例证,下面实施例中的化合物B能够在实施例中描述的体外条件下,持续480分钟释放CO,从本发明的意义上说这是很长的一段时间。
在根据该第二实施方案的一些化合物中,根据式(Ib)的杂合富马酸-一氧化碳释放分子在一侧被一种CO-释放羰基络合物取代,并且在另一侧被不同的CO-释放羰基络合物取代:
其中A-CORM和A’-CORM’如上面或下面式(I)中所定义。
有利地,CORM和CORM’分别为其中M为Mn(CO)4或Ru(CO)3Cl。
有利地,在式(Ib)的化合物中,A为Q-Z-W其中Z-W为-CH2-CH2-NR7-,其中R7代表(C1-C3)烷基,并且A’为Q’-Z’,其中Z’为-(C1-C6)烷基,优选为(C1-C3)烷基。
有利地,Q为O或NR2,优选为O,并且Q’为O或NR2,优选为O。
有利地,根据第二实施方案的化合物具有式(Ic),其中富马酸部分在两侧均被相同的CO-释放羰基络合物取代:
其中A-CORM如式(I)的化合物中所定义。
在该实施方案中,CORM优选代表选自以下的羰基金属络合物:
Mn(CO)5
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Co,Ru,Mn,Mo,V或Fe,优选代表Fe,Co,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价;
优选地,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Rh,Ru,Mn,Mo,V或Fe,优选代表Fe,Ru或Mn,以及
n为选定的整数,所述整数使金属M没有自由价;
甚至优选地,CORM代表选自以下的羰基金属络合物:
Mn(CO)5,
其中W代表O或NR4,其中R4代表-(C1-C6)烷基-,
L代表阴离子配体如卤素,或抗衡离子如BF4或PF6,
M代表Fe或Mn,以及
n为选定的整数,所述整数使金属M没有自由价。
一些有利的式(Ic)的化合物具有式(Ic1):
其中:
M代表Mn(CO)4或Ru(CO)3Cl,以及
Q和Z如式(I)中所定义。
有利地,在式(Ic1)的化合物中,Z-W为-CH2-CH2-NR7-,其中R7代表(C1-C3)烷基。
有利地,Q为O或NR2,优选为O。
一些其他的有利的式(Ic)的化合物具有式(Ic2):
一些进一步有利的式(Ic)的化合物具有式(Ic3):
其中:
Q和Z如式(I)中所定义。
有利地,Q-Z为氨基酸(如丝氨酸,半胱氨酸,酪氨酸或赖氨酸)的侧链。
进一步有利的式(Ic)的化合物具有式(Ic4):
其中:
Mx(CO)y代表Co2(CO)6或Co4(CO)10,优选代表Co4(CO)10,
Q和Z如式(I)中所定义。
更有利地,Q为O,并且Z为-(C1-C6)烷基,优选为(C1-C3)烷基。
有利地,式(I)的化合物选自:
在特别的实施方案中,式(I)的化合物为:
本发明还涉及一种药物组合物,所述药物组合物包含至少一种如之前所定义的式(I)的化合物,其药学上可接受的盐、溶剂化物或水合物,和至少一种药学上可接受的赋形剂。
本发明的药物组合物有利地适用于经由口服、舌下、皮下、肌内、静脉内、经皮、局部或直肠途径施用。还可以通过吸入,例如利用喷雾器施用本发明的药物组合物。可以通过与常规的药物载体(carrier)混合的用于施用的单位形式,向动物或向人施用活性成分。
当固体组合物制备为片剂形式时,将主要活性成分与药物载体(vehicle)和本领域技术人员已知的其他常规赋形剂混合。
可以以0.01mg至1000mg每日的剂量在药物组合物中使用本发明的化合物,每日一次仅施用一剂或每日施用几剂,例如每日施用两剂。每日施用的剂量有利地包含5mg至500mg,更有利地为10mg至200mg。然而,可以使用这些范围以外的剂量,本领域技术人员可以注意到所述剂量。
本发明进一步涉及式(I)的化合物,其药学上可接受的盐、溶剂化物或水合物,或者包含至少一种式(I)的化合物、其盐、溶剂化物或水合物的药物组合物,其作为药物的用途。
本发明进一步涉及至少一种式(I)的化合物,其药学上可接受的盐、溶剂化物或水合物,或者包含至少一种式(I)的化合物、其盐、溶剂化物或水合物的药物组合物,其在治疗心血管或炎性疾病中的用途。
本发明进一步涉及至少一种式(I)的化合物,其药学上可接受的盐、溶剂化物或水合物在制备用于治疗心血管或炎性疾病的药物中的用途。
本发明进一步涉及一种用于治疗心血管或炎性疾病的方法,其包括向需要其的人施用至少一种式(I)的化合物,其药学上可接受的盐、溶剂化物或水合物,或者施用包含至少一种式(I)的化合物、其药学上可接受的盐、溶剂化物或水合物的药物组合物。
根据本发明的炎性和心血管疾病包括例如心肌缺血和心脏疾病、类风湿性关节炎、急性和慢性皮肤伤口(伤口愈合)、炎症性肠病、术后肠梗阻、脑缺血、银屑病、糖尿病、糖尿病肾病、代谢综合征、镰状细胞病、神经变性疾病如阿尔茨海默氏病或帕金森氏病、神经性疼痛、高血压、肺动脉高压、败血症、脓毒性或内毒素性休克、出血性休克、多发性硬化、癌症和慢性阻塞性肺病。根据本发明的优选的炎性和心血管疾病为皮肤伤口(伤口愈合)、脑和心肌缺血、银屑病、糖尿病、多发性硬化、癌症和慢性阻塞性肺病。
本发明进一步涉及一种用于制备式(I)的化合物、其盐、溶剂化物或水合物的方法。
可以根据两种方法获得式(I)的化合物。
方法(a):
步骤(1):使用选自以下的式(II)的化合物将富马酸的二酰基氯化物、溴化物、氟化物或二活化酯,或者富马酸的单酯、单酰胺或单硫酯的单酰基氯化物、溴化物、氟化物或单活化酯酯化:
其中Z和Q如以上所定义。
可替代地,可以使用选自以下的式(III)的化合物将富马酸或富马酸的单酯烷基化:
其中Z和Q如以上所定义,Hal代表离去基如卤素或磺酸酯,如三氟甲磺酸酯。
步骤(2):使在步骤(1)中获得的化合物与合适的式L1L2Mx(CO)y的羰基金属络合物反应,在任选脱保护后生成式(I)的化合物,其中x为1或2并且y为1至10,并且L1和L2各自代表单齿配体或L1L2代表二齿配体。
因此,本发明涉及一种用于合成式(I)的化合物的方法,其包括式(IV)的富马酸衍生物:
其中:
X1代表如式(I)中所定义的A-R8,A-CORM,A’-CORM’或Q’-Y,以及
R8代表选自以下的基团:
与式L1L2Mx(CO)y的羰基金属络合物的反应,其中x为1或2并且y为1至10,L1和L2各自代表单齿配体或L1L2代表二齿配体。
方法(b):
使用式H-A-CORM的化合物将富马酸的二酰基氯化物、溴化物、氟化物或二活化酯,或者富马酸的单酯、单酰胺或单硫酯的单酰基氯化物、溴化物、氟化物或单活化酯酯化。
可替代地,可以使用式Hal-A-CORM的化合物将富马酸或富马酸的单酯烷基化,其中A如上面所定义,并且Hal代表离去基如卤素或磺酸酯,如三氟甲磺酸酯。
因此,本发明还涉及一种用于合成式(I)的化合物的方法,其包括式(V)的富马酸衍生物:
其中:
X1代表Cl,F,Br或酯,有利地代表活化酯,
X2代表Cl,F,Br或酯(有利地代表活化酯),如式(I)中所定义的A-CORM,A’-CORM’或Q’-Y,
与式H-A-CORM的化合物的反应,其中A-CORM如式(I)中所定义,
或者,
包括式(I)的化合物,其中X1和/或X2代表OH,与式Hal-A-CORM的化合物的反应,其中Hal代表离去基,如卤素或磺酸酯,如三氟甲磺酸酯。
从本发明的意义上说,术语“离去基”是指在亲核取代反应期间,可以容易地被亲核试剂替代的化学基团。
从本发明的意义上说,表述“活化酯”是指增强富马酸的羰基的反应性的酯。可以根据熟知的程序在反应前制备或原位生成这种活化酯。
可用于制备活化酯的试剂的示例包括偶联剂,如二异丙基碳二亚胺(DIC)、二环己基碳二亚胺(DCC)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)、羰二咪唑(CDI)、2H苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐(HBTU)、2-(lH-苯并三唑-1-基)-1,1,3,3-四甲基脲四氟硼酸盐(TBTU)、O(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐(HATU)或(苯并三唑-1-基氧基)三吡咯烷基磷六氟磷酸盐(PyBOP);任选与辅助偶联剂联合,如N-羟基-琥珀酰亚胺(NHS)、N-羟基-苯并三唑(HOBt)、3,4-二氢-3-羟基-4-氧代-1,2,3-苯并三唑(HOOBt)、l-羟基-7-氮杂苯并三唑(HAt)或N-羟基硫代琥珀酰亚胺(硫代NHS)。
当存在时,可以保护容易干扰期望的反应的官能团,例如杂原子,如N或O,以在合成程序期间避免不期望的反应。
本领域技术人员容易确定保护基是否是必要的。本领域已知并且例如在Greene的“Protective Groups In Organic synthesis”,(John Wiley&Sons,纽约(1981)中公开了合适的保护基。
可以通过富马酸或其衍生物与式H-Q’-Y的化合物的酯化、酰胺化或硫酯化,或者通过富马酸与式Hal-Y的化合物的烷基化,其中Hal为离去基如Br,Cl,I或OSO2CF3,并且Y如以上所定义,在常规条件下制备富马酸的单酯,即式(II)的化合物,其中X1代表Q’-Y。
可以根据上面描述的方法(b)通过将富马酸与式H-A-CORM或Hal-A-CORM的化合物反应,并且如需要将羧酸转化为酰基氯化物、溴化物、氟化物或活化酯,制备式(V)的化合物,其中X1代表A-CORM。
可以通过将富马酸的二酰氯与两个当量的式H-A-CORM的化合物或两个当量的选自以下的化合物反应:
然后根据富马酸酯的量与至少两个当量的式L1L2Mx(CO)y的羰基金属络合物反应,以一步程序获得式(Ic)的化合物,其中x为1或2,y为1至10,L1和L2各自代表单齿配体或L1L2代表二齿配体。
可以通过将富马酸酯的单酰基氯化物或富马酸的二酰基氯化物与合适量的阴离子锰羰基络合物如Na+[Mn(CO)5]-反应,以获得式(Ia2)和(1c2)的化合物。
根据方法(a)制备式H-A-CORM的化合物或式(I)的化合物的条件涉及本领域已知的方法和程序。
定义:
本发明仅涵盖稳定的化合物。在这点上,当提及“异构体”时,仅考虑稳定的异构体。
在说明书和权利要求书中定义的基团(group)、基团(radical)或片段(fragment)中,在括号中详细说明了碳原子数。例如,(C1-C6)烷基是指具有1至6个碳原子的烷基或基团。
在式中,是指连接至分子的其余部分的键。
对于包含两个或多个亚基的基团,使用“-”指示连接。例如,“-(C1-C6)烷基-芳基-(C1-C6)烯基-”是指连接至芳基基团的烷基基团,所述芳基基团自身连接至烯基,其中所述烷基和烯基连接至分子的其余部分。
从本发明的意义上说,表述“-(C1-C6)烷基”是指包含1至6个碳原子的非环的、饱和的、直线的或分支的烃链。-(C1-C6)烷基的示例包括甲基、乙基、丙基、丁基、戊基或己基。除非明确说明,丙基、丁基、戊基和己基的定义包括所有可能的异构体,特别是结构异构体。例如,丁基包括正丁基、异丁基、仲丁基和叔丁基。
从本发明的意义上说,表述“-(C2-C6)烯基”是指包含2至6个碳原子的非环的、饱和的、直线的或分支的烃链,至少两个所述碳原子经由双键相连。“-(C2-C6)烯基”的示例包括乙烯基(ethenyl)或乙烯基(vinyl)、丙烯基、丁烯基、戊烯基或己烯基。除非明确说明,丙烯基、丁烯基、戊烯基和己烯基的定义包括所有可能的异构体,特别是结构和/或位置异构体。
从本发明的意义上说,表述“-(C2-C6)炔基”是指包含2至6个碳原子的非环的、饱和的、直线的或分支的烃链,至少两个所述碳原子经由三键相连。“-(C2-C6)炔基”的示例包括乙炔基、丙炔基、丁炔基、戊炔基或己炔基。除非明确说明,丙炔基、丁炔基、戊炔基和己炔基的定义包括所有可能的异构体,特别是结构和/或位置异构体。
如本文所使用的术语取代的是指任何氢原子可以被取代基如氟替代。
从本发明的意义上说,表述“-(C3-C14)环烷基”是指包含3至14个碳原子的饱和或部分饱和的单、二或三环结构。“-(C3-C14)环烷基”的示例包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环戊烯基和环己烯基。
“-(C3-C8)环烷基”的示例包括环丙基、环丁基、环戊基、环己基、环庚基或环辛基。除非明确说明,环烷基可以被一个或多个基团如甲基、乙基、异丙基、羟基、氟、氯、溴和碘取代。
从本发明的意义上说,表述“-(C3-C8)杂环基”是指在环上具有3、4、5、6、7或8个原子的饱和杂环,其中1、2或3个选自N、O和S的杂原子替代了相应数量的碳原子。“-(C3-C8)杂环基”的示例包括吖丙啶基、环氧乙烷基、吡咯烷基、四氢呋喃基、噁唑基、哌啶基、哌嗪基和吗啉基。
术语“芳基”是指芳香单环,其可以与第二个饱和的、不饱和的或芳香环稠合。术语芳基包括但不限于以下示例,苯基、茚满基、茚基、萘基、蒽基、菲基、四氢萘基和二氢萘基。最优选的芳基为包含一个六元芳香环的那些。芳基可以被取代,优选被独立地选自烷基、烷氧基、卤素、羟基、氨基、硝基、氰基、三氟、羧酸或羧酸酯的一个或多个基团取代。
术语杂芳基是指如上面所定义的单或多环芳基,其中一个或多个碳原子已被一个或多个选自N、O和S的杂原子替代。除非明确说明,术语“杂芳基”包括所有可能的异构体,特别是位置异构体。
杂芳基的示例包括呋喃基、噻吩基、咪唑基、吡啶基、吡咯基、N-烷基吡咯基、嘧啶基、吡嗪基、四唑基、三唑基和三嗪基。杂芳基可以被取代,优选被独立地选自烷基、烷氧基、卤素、羟基、氨基、硝基、氰基、三氟、羧酸或羧酸酯的一个或多个基团取代。优选的杂芳基为在环上具有5或6个原子的那些,如吲哚基、吡咯基、吡啶基、吡唑基、三唑基、呋喃基或噻吩基。
如本文所使用,术语“卤素”是指氟、氯、溴或碘原子。
对于本发明的目的,术语“药学上可接受的”是指对于药物组合物的制备是有用的,并且对于药物用途通常是安全和无毒的。
在本发明的框架内,术语“药学上可接受的盐、水合物或溶剂化物”是指化合物的盐,其如以上所定义是药学上可接受的,并且其拥有相应化合物的药物活性。这种盐包含:
(1)水合物和溶剂化物,
(2)与无机酸如盐酸、氢溴酸、硫酸、硝酸和磷酸等形成的酸加成盐;或者与有机酸如乙酸、苯磺酸、富马酸、葡庚糖酸、葡萄糖酸、谷氨酸、乙醇酸、羟基萘甲酸(hydroxynaphtoic)、2-羟基乙磺酸、乳酸、马来酸、苹果酸、扁桃酸、甲磺酸、粘糠酸、2-萘磺酸、丙酸、琥珀酸、二苯甲酰基-L-酒石酸、酒石酸、对甲苯磺酸、三甲基乙酸和三氟乙酸等形成的酸加成盐,以及
(3)当存在于化合物中的酸质子被金属离子,如碱金属离子、碱土金属离子、或铝离子替代所形成的盐;或者与有机碱或无机碱配位所形成的盐。可接受的有机碱包含二乙醇胺、乙醇胺、N-甲基葡萄糖胺、三乙醇胺、氨丁三醇等。可接受的无机碱包含氢氧化铝、氢氧化钙、氢氧化钾、碳酸钠和氢氧化钠。
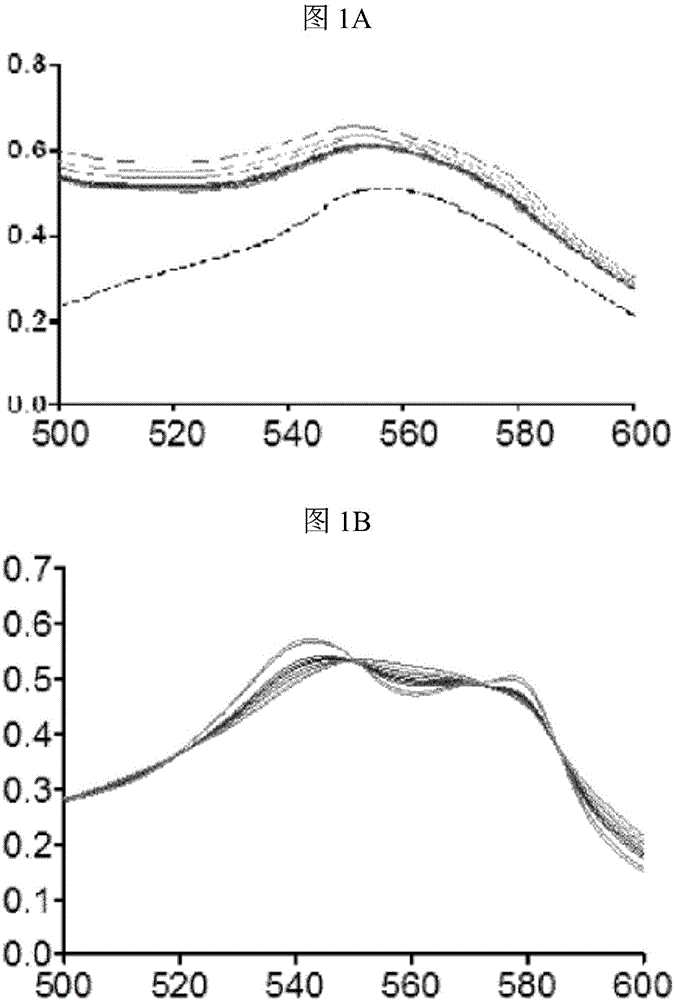
附图说明
图1表示在不同时间,在根据WO 2012/076696的姜黄素衍生物(图1A)、化合物A(图1B)和化合物B(图1C)的存在下,一氧化碳肌红蛋白(MbCO)的UV吸收。在540和578nm处的吸收峰的存在表明了一氧化碳肌红蛋白(MbCO)的形成(图1A-C)。在图1D中提供不同浓度的化合物C根据实施例2的COP-1测试的细胞内CO。
图2表示不存在(CON)或存在升高浓度的化合物A(图2A),或化合物C、WO 2008/003953的对比化合物CORM-401和对比化合物J(图2B)时,在BV2小胶质细胞中的血红素加氧酶活性。血红素加氧酶活性作为对照的百分比表示在Y轴上。
图3在Y轴上表示在BV2小胶质细胞中(CON),在使用脂多糖(LPS,1μg/ml)(LPS)处理24h的BV2小胶质细胞中,以及在升高浓度的化合物A(图3A)或化合物B(图3B)的存在下使用脂多糖(LPS,1μg/ml)(LPS)处理的BV2小胶质细胞中以μM表示的亚硝酸盐生成量。图3C和图3D在Y轴上表示在BV2小胶质细胞中(CON),在使用脂多糖(LPS,1μg/ml)处理24h的BV2小胶质细胞中,以及在升高浓度的化合物C(1,2.5和5μM)的存在下,和在5μM的根据WO 2012/076696的姜黄素衍生物的存在下(图3C),或在5μM的对比富马酸衍生物(化合物J)和WO 2008/003953的对比化合物CORM-401的存在下(图3D)使用脂多糖(LPS,1μg/ml)(LPS)处理的BV2小胶质细胞中以μM表示的亚硝酸盐生成量。
图4表示随着化合物A或B浓度升高(X轴),化合物A(图4A)、化合物B(图4B)在BV2小胶质细胞中测量的细胞毒性,根据WO 2012/076696的对比姜黄素衍生物(图4C)在RAW巨噬细胞中测量的细胞毒性,化合物A在角化细胞(图4D)中测量的细胞毒性,或化合物A在THP-1细胞(图4E)中测量的细胞毒性相比对照的百分比(Y轴)。
图5表示在BV2小胶质细胞中(对照),在存在化合物A的BV2小胶质细胞中,以及在存在化合物B的BV2小胶质细胞中Nrf-2的表达(图5A)。通过“+”指示实验中存在化合物,通过“-”指示实验中不存在化合物。图5B表示在不存在(对照)或存在升高浓度的化合物A的BV2小胶质细胞中HO-1的表达。图5C表示在升高浓度的化合物C、WO 2008/003953的对比化合物CORM-401或对比化合物J的存在下,BV2小胶质细胞中HO-1的表达和Nrf-2的表达。图5D表示在化合物C的存在下,在角化细胞中随着时间的HO-1的表达和Nrf-2的表达。图5E表示在升高浓度的已知活化Nrf2的化合物C或对比富马酸二甲酯(DMF)或对比阳性对照C+(肉桂醛,100μM)的存在下,在角化细胞中HO-1的表达和Nrf-2的表达。图5F表示在升高浓度的化合物A、化合物B或化合物C的存在下,在THP-1细胞中HO-1的表达和Nrf-2的表达。
图6表示在化合物A(图6A,-■-)和化合物B(图6A和图6B,-●-)的存在下,使用脱氧肌红蛋白(Mb,100μM)的随时间(X轴,以分钟表示)而变化的释放的CO的浓度(Y轴,以μM计的检测的一氧化碳肌红蛋白的量表示)。
图7表示在化合物C和化合物E的存在下,在用LPS处理的人巨噬细胞中的TNF-α生成量。
图8表示在化合物C和化合物E的存在下,在用LPS处理的人THP-1细胞中的TNF-α生成量。
具体实施方式
通过以下非限制性实施例阐明本发明。
实施例
实施例1:根据本发明的杂合富马酸-CO-RM的合成
化合物A:
步骤1:制备:
将富马酸单乙酯(1.544g,10.78mmol)、溴丙炔(4.488g,37.73mmol)和碳酸钾(1.851g,13.34mmol)在丙酮(104mL)中的溶液在室温搅拌48小时。将所形成的溴化钾过滤掉,并减压去除溶剂。通过闪式色谱法,在硅胶上用环己烷/乙酸乙酯(8/2Rf=0.5)洗脱来纯化粗混合物,以68%的产率获得作为无色液体的期望的产物。
在CDCl3中的NMR(400MHz):
1Hδ(ppm):1.32(t,3H,CH3,J=7.1Hz);2.52(t,1H,CH,J=0.1Hz);4.26(q,2H,CH2,J=7.1Hz);4.8(d,2H,CH2,J=2.4Hz);6.86(d,1H,CH=CH,J=15.8Hz);6.92(d,1H,CH=CH,J=15.8Hz)。
13Cδ(ppm):14.2(CH3-CH2);52.8(CH2O);61.6(CH3-CH2);75.6(CH);76.8(C);132.5(CH=CH);134.9(CH=CH);164.3(C=O);164.8(C=O)。
元素分析:计算值:C,59.34;H,5.53。实测值:C,59.13;H,5.53.。
MS(EI)m/z:[M+H]+=183(17);137(25.44);127(100);99.1(29.44);81.1(17.05);79.1(24.25);71.1(17.94);55(48.89);53.1(31.65)。
IR(ATR,cm-1):3300,1663,1617。
步骤2:化合物A的制备
在避光和氩气惰性气氛保护下的施伦克(Schlenk)管中,将步骤1中获得的富马酸酯(0.1012g,0.5560mmol,1当量)、八羰基二钴(Co2(CO)8,0.1901g,0.5560mmol,1当量)在无水乙醚(2mL)中的溶液在室温搅拌3h40(通过TLC监测反应的完成)。避光减压去除溶剂。将粗混合物溶于环己烷(15mL)中,并将沉淀的KBr过滤至硅藻土上。在蒸发环己烷后,获得油状紫黑色残余物,并通过氧化铝制备型色谱(洗脱剂:戊烷/乙醚:9/1)将其纯化。
获得187.4mg的油状化合物。将101mg的油在-18℃在已烷中重结晶获得70mg作为红黑色晶体的化合物A。
在CDCl3中的NMR(400MHz)
1Hδ(ppm):1.26(s,3H,CH3);4.19(s,2H,CH2O);5.35(s,2H,CH2);6.02(s,1H,CH);6.85(s,2H,CH=CH)。
13Cδ(ppm):13.1(CH3-CH2);60.4(OCH2);64.8(CH3-CH2);70.9(CH);86.7(C);131.6(CH=CH);133.6(CH=CH);163.3(C=O);163.8(C=O);197.9(C=O)。
元素分析:计算值:C,38.49;H,2.15。实测值:C,38.45;H,2.25。
IR(ATR,cm-1):2100,2050(CO);1967(CO);1717。
化合物B:
步骤1:制备:
在施伦克管中,向溶解于10mL氯仿中的0.1g(1当量,7×10-4mol)碳酸钾和0.275g(2当量,1.4×10-3mol)溴化四乙胺(Et4NBr)中依次加入76μl(1当量,7×10-4mol)富马酰氯和82μl(2当量,1.4×10-3mol)丙炔醇。然后将溶液在室温搅拌过夜。
减压去除溶剂,在硅胶闪式色谱(洗脱剂:8/2环己烷/乙酸乙酯)后,以50%的产率获得作为白色固体的产物。
1H NMR(CDCl3):2.46(2H,3Hz,t,H1),4.74(4H,3Hz,t,H3),6.87(2H,s,H5)
13C NMR(CDCl3):75.6(C3),133.7(C5),163.8(C4)
IR:ν=1650,1720cm-1
步骤2:化合物B的制备
在避光的施伦克管中,将200mg(1.04×10-3mol)在步骤1中获得的富马酸酯溶解于10ml氯仿中。在室温滴加八羰基二钴的氯仿溶液(356mg,1.04×10-3mol在15ml氯仿中),并通过TLC监测反应进程。在反应完成后,减压去除氯仿,在硅胶色谱(洗脱剂6/4环己烷/氯仿)后获得作为红黑色粉末的产物。
1H NMR(CDCl3):6.95(1H,H5),6.04(1H,H1),5.40(2H,H3)
13C NMR(CDCl3):199(12C,C6),164.6(2C,C4),133,(2C,C5),87.9(2C,C2),72(2C,C1),66.2(2C,C3)
元素分析:计算值:C,34.58;H,1.06;发现值:C,34.99;H,1.30。
IR:ν=2097,2013,1714cm-1
化合物C:
步骤1:(2-羟乙基)(甲基)氨基甲酰二硫代酸钠的制备
在0℃,在氩气惰性气氛下,向二硫化碳(452μL,7.5mmol,1.5当量)在10mL无水THF中的溶液中滴加2-(甲基氨基)乙醇(400μL,5.0mmol,1.0当量)。在0℃搅拌5分钟后,分批加入氢化钠(在矿物油中的60%分散体,200mg,5.0mmol,1.0当量)。在0℃搅拌30分钟后,减压浓缩混合物,用环已烷洗涤以去除矿物油,并减压干燥过夜以获得淡黄色粉末(842mg,4.9mmol,产率97%)。
1H NMR(400MHz,MeOD)δ(ppm):3.57(s,3H,CH3),3.87(t,J=6Hz,2H,CH2N),4.27(t,J=6Hz,2H,CH2O)
13C NMR(100MHz,MeOD)δ(ppm):44.7(CH3N),59.1(CH2N),61.2(CH2O),214.0(CS2)。
步骤2:制备
向Mn(CO)5Br(137mg,0.50mmol,1.0当量,根据US 2010/0105770制备)的氩气吹洗的甲醇(8mL)溶液中加入在步骤1中制备的(2-羟乙基)(甲基)氨基甲酰二硫代酸钠(87mg,0.50mmol,1.0当量)。在氩气惰性气氛下,在45℃搅拌3h30后,将混合物溶解于乙醚中,在硅藻土上过滤并浓缩以获得黄色油(149mg,0.47mmol,产率94%),其不作进一步纯化而在步骤3中使用。
1H NMR(400MHz,CDCl3)δ(ppm):3.34(s,3H,CH3N),3.48(s,1H,OH),3.92(s,4H,CH2N,CH2O)
13C NMR(100MHz,CDCl3)δ(ppm):38.5(CH3N),53.4(CH2N),60.3(CH2O),198.7(CS2),208.9(CO)
55Mn NMR(100MHz,CDCl3)δ(ppm):-993.1
步骤3:化合物C的制备
在氩气惰性气氛下,向在步骤2中获得的化合物(149mg,0.47mmol,1.0当量)的无水二氯甲烷(10mL)溶液中加入(E)-乙基-4-氯-4-氧代丁-2-烯酸酯(115mg,0.71mmol,1.5当量,如J.Am.Chem.Soc.,1996,118,8266-8277中描述所制备)和三乙胺(99μL,0.71mmol,1.5当量)。在氩气中在20℃搅拌18小时后,将混合物减压浓缩。将粗混合物溶解于乙醚中,并在硅藻土上过滤。在蒸发溶剂和硅胶闪式色谱(洗脱剂:乙酸乙酯/环已烷:3/7+0.1%NEt3)后,获得107mg黄色固体(0.24mmol,产率51%)。
1H NMR(400MHz,CDCl3)δ(ppm):1.33(s,3H,CH3C),3.30(s,3H,CH3N),4,05(s,2H,CH2N),4.27(s,2H,CH2O),4,46(s,2H,CH2O),6.87(s,1H,CH=C),6.88(s,1H,CH=C)
13C NMR(100MHz,CDCl3)δ(ppm):14.1(CH3C),37.8(CH3N),49.8(CH2N),61.4(CH2O),61.5(CH2O),132.4(CH=C),134.9(CH=C),164.6(CO酯),164.7(CO酯),197.0(CS2),210.1(CO金属)
55Mn NMR(100MHz,CDCl3)δ(ppm):-984.5
元素分析:计算值C(37.93),H(3.18),N(3.16);实测值C(38.12),H(3.28),N(3.09)。
化合物D:
步骤1:化合物AO234的制备
向[RuCl2(CO)3]2(324mg,0.63mmol,0.5当量)在甲醇(13mL)中的混悬液中加入(2-羟乙基)(甲基)氨基甲酰二硫代酸钠(如在合成化合物C的步骤1中所制备)(219mg,1.26mmol,1.0当量)。在氩气中在20℃搅拌3h后,将反应混合物真空浓缩,溶解于EtOAc中,在硅藻土上过滤并真空浓缩以获得作为浅黄色固体的化合物AO234(435mg,1.17mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ3.96-3.77(M,4H,CH2N,CH2O),3.37(s,3H,CH3N)。13C NMR(100MHz,MeOD)δ210.27(CS2),188.95(CO金属),188.72(CO金属),59.81(CH2O),55.03(CH2N),38.86(CH3N)。
步骤2:化合物D的制备
在氩气中,向化合物AO234(435mg,1.17mmol,1.0当量)的无水二氯甲烷(16mL)溶液中加入(E)-4-氯-4-氧代-2-丁烯酸乙酯(285mg,1.76mmol,1.5当量)和4-DMAP(215mg,1.76mmol,1.5当量)。在氩气中在40℃搅拌17h后,将反应混合物真空浓缩。将所得的粗产物通过硅胶柱色谱用环已烷/EtOAc(60/40)洗脱纯化,并用叶片泵干燥一夜,以50%的产率(两步)获得作为白色固体的化合物D(312mg,0.63mmol)。1H NMR(400MHz,CDCl3)δ6.89(d,J=21.2Hz,1H,CH=C),6.85(d,J=21.2Hz,1H,CH=C),4.48(m,2H,CH2O),4.30(m,1H,CH2N),4.26(q,J=7.2Hz,2H,CH2O),3.80(m,1H,CH2N),3.35(s,3H,CH3N),1.32(t,J=7.2Hz,3H,CH3C)。13C NMR(100MHz,CDCl3)δ211.09(CS2),186.98(CO金属),186.67(CO金属),186.66(CO金属),164.59(CO酯),164.47(CO酯),135.07(CH=C),132.17(CH=C),61.51(CH2O),61.13(CH2O),50.09(CH2N),37.92(CH3N),14.05(CH3C)。分析计算值C13H14ClNO7RuS2(496.91):C,31.42;H,2.84;N,2.82。实测值:C,31.49;H,2.80;N,2.79。
化合物E:
步骤1:化合物AO 83的制备
在氩气中在0℃,向1-(2-羟基乙基)哌嗪(651mg,5.00mmol,1.0当量)的无水四氢呋喃(15mL)溶液中滴加二硫化碳(452μL,7.50mmol,1.5当量)。在0℃搅拌15min后,分批加入氢化钠(在矿物油中的60%分散体)(200mg,5.00mmol,1.0当量)。在0℃搅拌40min后,将反应混合物真空浓缩。使用环已烷将所得的粗产物洗涤几次以去除矿物油,然后真空浓缩并用叶片泵干燥一夜,以96%的产率获得作为淡黄色粉末的化合物AO83(1.10g,4.82mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ4.46(t,J=4.8Hz,4H,CH2N),3.71(t,J=5.9Hz,2H,CH2O),2.55(t,J=5.1Hz,6H,CH2N)。13C NMR(100MHz,MeOD)δ60.98(CH2N),59.80(CH2O),54.35(CH2N),51.20(CH2N)。
步骤2:化合物AO208的制备
向化合物AO83(318mg,1.39mmol,1.1当量)的甲醇(20mL)溶液中加入Mn(CO)5Br(346mg,1.26mmol,1.0当量)。在氩气中在45℃搅拌3h后,将反应混合物真空浓缩,溶解于乙醚中,在硅藻土上过滤并真空浓缩,以94%的产率获得作为黄色油的化合物AO208(443mg,0.32mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ3.91(s,4H,CH2N),3.68(s,2H,CH2O),2.58(s,6H,CH2N)。13C NMR(100MHz,MeOD)δ207.67(CO金属),60.60(CH2N),59.79(CH2O),53.34(CH2N),46.86(CH2N)。55Mn NMR(100MHz,MeOD)δ-1025.7。分析计算值C11H13MnN2O5S2(372.30):C,35.49;H,3.52;N,7.52。实测值:C,35.64;H,3.71;N,7.10。
步骤3:化合物E的制备
在氩气中,向化合物AO208(247mg,0.66mmol,1.0当量)的无水二氯甲烷(9mL)溶液中加入(E)-4-氯-4-氧代-2-丁烯酸乙酯(162mg,1.00mmol,1.5当量)和4-DMAP(122mg,1.00mmol,1.5当量)。在氩气中在40℃搅拌18h后,将反应混合物真空浓缩,然后溶解于乙醚中,在硅藻土上过滤并真空浓缩。将所得的粗产物通过硅胶柱色谱用环已烷/EtOAc(70/30+0.1%NEt3)洗脱纯化,以45%的产率获得作为黄色固体的化合物E(151mg,0.30mmol)。1H NMR(400MHz,C6D6)δ6.96(s,2H,CH2=C),3.88(s,2H,CH2O),3.79(s,2H,CH2O),3.23(s,4H,CH2N),1.87(s,2H,CH2N),1.65(s,4H,CH2N),0.86(s,3H,CH3C)。13C NMR(100MHz,C6D6)δ206.93(CO金属),164.55(CO酯),134.27(CH2=C),133.33(CH2=C),61.82(CH2O),61.23(CH2O),55.78(CH2N),51.54(CH2N),45.87(CH2N),13.95(CH3C)。55Mn NMR(100MHz,C6D6)δ–990.3。分析计算值C17H19MnN2O8S2(498.41):C,40.97;H,3.84;N,5.62。实测值:C,41.08;H,4.06;N,5.73。
化合物F:
步骤1:化合物AO228的制备
向[RuCl2(CO)3]2(256mg,0.50mmol,0.5当量)的甲醇(10mL)混悬液中加入化合物AO83(228mg,1.00mmol,1.0当量)。在氩气中在20℃搅拌4h后,将反应混合物真空浓缩,溶解于EtOAc中,在硅藻土上过滤并真空浓缩,以87%的产率获得作为浅黄色固体的AO228(370mg,0.87mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ3.96(d,J=5.0Hz,4H,CH2N),3.72(t,J=5.6Hz,2H,CH2O),2.76(s,4H,CH2N),2.68(s,2H,CH2N)。13C NMR(100MHz,MeOD)δ188.76(CO金属),188.59(CO金属),60.50(CH2N),59.41(CH2O),52.98(CH2N),47.00(CH2N)。
步骤2:化合物F的制备
在氩气中,向化合物AO228(370mg,0.87mmol,1.0当量)的无水二氯甲烷(11mL)溶液中加入(E)-4-氯-4-氧代-2-丁烯酸乙酯(211mg,1.30mmol,1.5当量)和4-DMAP(159mg,1.30mmol,1.5当量)。在氩气中在40℃搅拌16h后,将反应混合物真空浓缩,然后溶解于EtOAc中,在硅藻土上过滤并真空浓缩。将所得的粗产物通过硅胶柱色谱用环已烷/EtOAc(50/50)洗脱纯化,并用叶片泵干燥一夜,以40%的产率获得作为白色固体的化合物F(193mg,0.35mmol)。1H NMR(400MHz,CDCl3)δ6.87(s,2H),4.34(t,J=5.6Hz,2H),4.27(q,J=7.1Hz,2H),3.88(t,J=4.9Hz,4H),2.75(t,J=5.5Hz,2H),2.65(m,4H),1.33(t,J=7.1Hz,3H)。13C NMR(100MHz,CDCl3)δ186.78(CO金属),164.82(CO酯),134.26(CH2=C),133.07(CH2=C),62.09(CH2O),61.47(CH2O),56.06(CH2N),51.88(CH2N),46.48(CH2N),14.11(CH3C)。分析计算值C16H19ClN2O7RuS2(551.98):C,34.81;H,3.47;N,5.08。实测值:C,35.04;H,3.59;N,4.91。
化合物G:
在20℃,在氩气下,在施伦克管中,在剧烈搅拌下向汞(240μL)中分批加入钠(30mg,1.28mmol,2.8当量)。在钠消失后,引入无水四氢呋喃(5mL),然后加入十羰基二锰(decacarbonyldimanganese)(179mg,0.46mmol,1.0当量)。在20℃剧烈搅拌3h并倾析10min后,通过套管将上层转移至另一个施伦克管中并冷却至-78℃,然后滴加(E)-4-氯-4-氧代-2-丁烯酸乙酯(242mg,1.00mmol,2.2当量)。在-78℃搅拌1h并在20℃搅拌18h后,将混合物真空浓缩。将粗产物溶解于已烷(6mL)中,在20℃搅拌30min,在熔结玻璃(frit glass)上过滤,将滤液储存于-18℃持续18h以结晶。通过去除溶剂收集结晶的固体,并用叶片泵短暂干燥,以5%的产率获得作为橙红色固体的化合物G(29mg,0.090mmol)。1H NMR(400MHz,C6D6)δ7.40(d,J=15.5Hz,1H),5.87(d,J=15.3Hz,1H),3.90(q,J=7.2Hz,2H),0.87(t,J=7.2Hz,3H)。55Mn NMR(100MHz,C6D6)δ-1779.7。分析计算值C11H7MnO8(322.11):C,41.02;H,2.19。实测值:C,40.76;H,2.50。
化合物I:
步骤1:化合物AO219的制备
在0℃在氩气中,向2-(甲基氨基)乙醇(400μL,5.00mmol,1.0当量)的氯仿(50mL)溶液中加入亚硫酰氯(1.09mL,15.0mmol,3.0当量)。在50℃搅拌17h后,将溶剂浓缩至约10mL,然后加入乙醚(40mL)以沉淀化合物,将其收集在熔结玻璃上,使用乙醚洗涤几次并用叶片泵干燥一夜,以87%的产率获得作为白色固体的化合物AO219(567mg,4.36mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ3.91(t,J=5.4Hz,2H,CH2Cl),3.42(t,J=5.4Hz,2H,CH2N),2.77(s,3H,CH3N)。13C NMR(100MHz,MeOD)δ51.46(CH2N),40.22(CH2Cl),33.64(CH3N)。
步骤2:化合物AO220的制备
向化合物AO219(567mg,4.36mmol,1.0当量)的水(20mL)溶液中加入叠氮化钠(850mg,13.1mmol,3.0当量)。在80℃搅拌18h后,加入氢氧化钠溶液(5%)(5mL)。用乙醚萃取反应混合物(×3)。将有机层用硫酸镁干燥,过滤,用HCl(气体)处理,浓缩并用叶片泵干燥5h,以80%的产率获得作为白色固体的化合物AO220(476mg,3.48mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ3.75(t,J=5.6Hz,2H,CH2N),3.17(t,J=5.6Hz,2H,CH2N3),2.73(s,3H,CH3N)。13C NMR(100MHz,MeOD)δ49.05(CH2N),48.29(CH2N3),33.66(CH3N)。
步骤3:化合物AO222的制备
向CuSO4.5H2O(864mg,3.46mmol,1.0当量)的水(40mL)溶液中加入炔丙醇(propargylic alcohol)(313μL,5.19mmol,1.5当量)和抗坏血酸钠(685mg,3.46mmol,1.0当量)。搅拌20min后,向反应混合物中加入化合物AO220(473mg,3.46mmol,1.0当量)的水(20mL)溶液。在50℃搅拌24h后,在玻璃粉(glass frit)上过滤反应混合物。将滤液用二氯甲烷洗涤,然后通过加入KOH调节pH至10。过滤后,真空浓缩水溶液并用二氯甲烷萃取几次。将有机层用硫酸镁干燥,过滤,浓缩并用叶片泵短暂干燥,以68%的产率获得作为白色固体的化合物AO222(366mg,2.34mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,CDCl3)δ7.63(s,1H,CH=C),4.80(s,2H,CH2O),4.46(t,J=5.8Hz,2H,CH2N),3.08(t,J=5.8Hz,2H,CH2N),2.45(s,3H,CH3N),1.61(se,2H,OH,NH)。13C NMR(100MHz,CDCl3)δ147.46(C=C),122.26(CH=C),56.73(CH2O),51.04(CH2N),50.01(CH2N),36.00(CH3N)。
步骤4:化合物AO224的制备
在氩气中在0℃,向化合物AO222(366mg,2.34mmol,1.0当量)的无水四氢呋喃(25mL)溶液中滴加二硫化碳(212μL,3.51mmol,1.5当量)。在0℃搅拌5min后,分批加入氢化钠(在矿物油中的60%分散体)(94mg,2.34mmol,1.0当量)。在0℃搅拌30min后,将反应混合物真空浓缩。将所得的粗产物溶解于甲醇/环己烷(1/1)的混合物中。在分层后,将甲醇层用环己烷洗涤并真空浓缩。将胶状固体溶解于最少量的二氯甲烷中,用乙醚稀释,真空浓缩并用叶片泵干燥一夜,以69%的产率获得作为棕色固体的化合物AO224(444mg,1.75mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ7.93(s,1H,CH=C),4.68(s,2H,CH2O),4.51(t,J=6.2Hz,2H,CH2N),3.04(t,J=6.2Hz,2H,CH2N),2.39(s,3H,CH3N)。13C NMR(100MHz,MeOD)δ149.10(C=C),124.51(CH=C),56.48(CH2O),51.76(CH2N),50.50(CH2N),35.74(CH3N)。
步骤5:化合物AO225的制备
向化合物AO224(444mg,1.75mmol,1.0当量)的甲醇(30mL)溶液中加入Mn(CO)5Br(487mg,1.75mmol,1.0当量)。在氩气中在45℃搅拌3h后,将反应混合物真空浓缩,溶解于乙酸乙酯中,在硅藻土上过滤并真空浓缩,以67%的产率获得作为黄色油的化合物AO225(464mg,1.16mmol),其不作进一步纯化而在下一步中使用。1H NMR(400MHz,MeOD)δ7.93(s,1H,CH=C),4.75(t,J=5.7Hz,2H,CH2N),4.67(s,2H,CH2O),4.26(t,J=5.6Hz,2H,CH2N),3.19(s,3H,CH3N)。13C NMR(100MHz,MeOD)δ210.99(CO金属),177.03(CS2),149.30(C=C),124.76(CH=C),56.50(CH2O),51.63(CH2N),47.86(CH2N),37.31(CH3N)。55Mn NMR(100MHz,MeOD)δ–1019.2。
步骤6:化合物I的制备
在氩气中,向化合物AO225(462mg,1.16mmol,1.0当量)的无水二氯甲烷(15mL)溶液中加入(E)-4-氯-4-氧代-2-丁烯酸乙酯(282mg,1.74mmol,1.5当量)和4-DMAP(213mg,1.74mmol,1.5当量)。在氩气中在40℃搅拌16h后,将反应混合物真空浓缩。将所得的粗产物用环已烷洗涤,干燥,溶解于最少量的二氯甲烷中,并用环已烷稀释以使其沉淀。在过滤和干燥后,用乙酸乙酯萃取残余物(×6)。将有机层在硅藻土上过滤,真空浓缩,用叶片泵干燥,并在石油醚和乙酸乙酯的混合物中结晶,以25%的产率获得作为黄色固体的化合物。1H NMR(400MHz,CDCl3)δ6.96(d,J=16.0Hz,1H,CH=C),6.91(d,J=15.9Hz,1H,CH=C),6.81(s,1H,CH=C),5.12(s,2H,CH2O),3.86(q,J=7.2Hz,2H,CH2O),3.53(t,J=6.2Hz,2H,CH2N),3.11(t,J=6.1Hz,2H,CH2N),2.14(s,3H,CH3N),0.85(t,J=7.2Hz,3H,CH3C)。13C NMR(100MHz,CDCl3)δ205.06(CO金属),164.75(CO酯),164.74(CO酯),143.09(C=C),134.65(CH=C),132.88(CH=C),124.55(CH=C),61.14(CH2O),58.38(CH2O),50.24(CH2N),45.85(CH2N),36.98(CH3N),13.92(CH3C)。55Mn NMR(100MHz,CDCl3)δ–1011.8。
实施例2:本发明化合物的CO-释放的评价
在肌红蛋白氧化测试中化合物A和B的CO-释放的评价:
在透明容器(cuvette)中,用1×磷酸盐缓冲盐水(PBS,pH 7.4)从500μM的储备液制备肌红蛋白(冻干的马心脏;终浓度44μM),并通过加入连二亚硫酸钠将肌红蛋白转化为脱氧肌红蛋白(deoxyMb)。在整个测试中,在分光光度计室中使用JASCO温度控制器将溶液维持在37℃。记录deoxyMb光谱,其揭露了在560nm处的特征峰。然后,将2倍过量的CO-RM分子加入至透明容器中,以获得最大一氧化碳-肌红蛋白(Mb-CO)光谱(饱和Mb-CO)。如对Mb-CO光谱所预期,观察到了542和576nm处的典型吸收峰。通过将化合物加入至44μM的deoxyMb中,并用400μl橄榄油覆盖溶液以防止CO损失或氧气(其与CO竞争结合肌红蛋白)扩散,测量来自化合物A、化合物B(30μM)或以下式:
的根据WO 2012/076696的姜黄素-CO-RM(30μM)的CO的释放。在加入化合物后和在连续的时间点上立即扫描样品,以监测Mb-CO的形成(μM)。
使用4点非线性回归法校正原始的Mb-CO数据,以补偿由于CO-释放化合物造成的吸收中的移位。使用如之前所描述的在540nm处的吸光度值和来自饱和Mb-CO光谱的消光系数,计算在每个时间点上Mb-CO的浓度。5用Mb-CO浓度(μM)相对时间(min)作图,使用GraphPad Prism软件5.0版(圣迭戈,加利福尼亚州),通过应用玻尔兹曼(Boltzmann)S形非线性回归曲线拟合确定t1/2。t1/2表示Mb-CO的浓度等于在测试条件下形成的最大Mb-CO的一半所采用的时间。
在540和578nm处的吸收峰的存在指示了通过由CO-RM分子释放的一氧化碳的MbCO的形成。
结果示于图1A-C中。
如这些结果所示,化合物A和化合物B释放一氧化碳,而根据WO 2012/076696的姜黄素衍生物不释放一氧化碳。
对于化合物A和B,还测量了随时间而变化的释放的CO的量。结果示于图6中。
结果表明相比于化合物B,化合物A以较快的速度放出CO,但是对于化合物B,由于两个双金属羰基络合物的存在,最终放出的CO的量要高得多。
因此可以推断出相比于式(Ic)的双金属杂合物,式(Ia)的单金属杂合物以较快的速度释放CO,但是对于化合物(Ic),由于两个双金属羰基络合物的存在,最终释放的CO的量要高得多。因此,式(Ia)的化合物对于快的生物活性(突释效应)是有利的。然而,由于式(Ic)的双金属杂合物能够释放更多的CO持续较长的一段时间,其对于持久的生物活性是有利的。
在COP-1测试中,暴露于化合物C的BV2小胶质细胞中的CO的探测
为了探测CO释放,将BV2小胶质细胞(106细胞/ml)洗涤一次,用含有Ca2+和Mg2+的PBS温育,然后在37℃避光下用化合物C和下式的根据WO 2008/003953的对比化合物CORM-401(5μM)处理15min:
对比化合物CORM 401。
然后,在37℃避光下将细胞装载在CO敏感的荧光探针COP-1(0.001mM)上持续30min。使用流式细胞仪,将结果表示为校正的平均荧光强度(MFIc)。MFIc=MFI染色处理的细胞–MFICOP-1背景。
结果示于图1D中。
如这些结果所示,化合物C比根据WO 2008/003953的对比化合物CORM-401释放显著更多的一氧化碳。
这些结果证明,本发明的化合物的富马酸部分引起细胞内(in cellulo)对一氧化碳释放的积极作用。此外这些结果证明,富马酸部分与CO-释放分子(CORM)的组合对一氧化碳释放具有协同作用。
实施例3:化合物A和B的生物活性的评价:
实施例3.1a:化合物A和B的血红素加氧酶活性的评价
通过分光光度测试确定血红素加氧酶活性,由于464和530nm之间吸光度的不同,所述分光光度测试测量胆红素的形成(胆红素的消光系数40mM-1·cm-1)。用升高浓度的化合物A温育BV2小胶质细胞6h。温育后,将细胞收集至含有2mM MgCl2的冰冷PBS(pH 7.4)中,并储存在-80℃。在含有PBS/MgCl2、作为血红素加氧酶底物的20μM氯高铁血红素(Frontier Scientific,Newark,DE)、作为胆绿素还原酶来源的大鼠肝细胞溶质、2mM葡萄糖-6-磷酸、0.5U/ml葡萄糖-6-磷酸脱氢酶和0.8mM NADPH的反应混合物中,将细胞样品在37℃避光温育1h。通过加入1ml纯氯仿终止反应,并通过涡旋离心循环提取仅含有胆红素的溶液。使用UltraspecTM 500pro可见光分光光度计(GE Healthcare Life Sciences)获得对应于胆红素的形成的吸光度值,数据显示为皮摩尔胆红素/mg蛋白质/h。使用Pierce BCA蛋白质测试试剂盒(Thermo Scientific)测量蛋白质浓度。
结果示于图2A中。
结果显示,化合物A在低至5μM的浓度显著提高HO-1活性。
实施例3.1b:化合物C的血红素加氧酶活性的评价
如上面所描述确定化合物C的血红素加氧酶活性,并将其与对比化合物CORM-401和对比富马酸衍生物(化合物J)作比较:
结果示于图2B中。
这些结果显示,化合物C在低至1μM的浓度显著提高HO-1活性。此外,相比于对比化合物J(其为不包含任何CORM的富马酸衍生物),化合物C显示出改进的HO-1活性。因此这证明,富马酸部分与CO-释放分子(CORM)的组合对血红素加氧酶活性具有协同作用。
实施例3.2:化合物A、化合物B和化合物C对炎症的作用:亚硝酸盐生成量的测定
在已用脂多糖(LPS,1μg/ml)处理24h的BV2细胞中,在存在或不存在升高浓度的化合物A或化合物B时,测量亚硝酸盐生成量(一种炎症指标)。在温育结束时,使用格里斯(Griess)法测量亚硝酸盐水平。12,14简言之,将细胞板以10,000×g离心5min,在室温将不含细胞的上清液用相同体积的格里斯试剂(2mM N-(1-萘基)乙烯肼(N-(1-naphtyl)ethylenediamide)、30mM磺胺和140mM正磷酸(o-phosphoric acid))温育10min。在560nm处测量吸光度。由亚硝酸钠的标准曲线计算亚硝酸盐的浓度(μM)。
结果示于图3中。
化合物A明显降低LPS介导的亚硝酸盐积累,这显示Co2(CO)6基团有助于亚硝酸盐水平的下降。令人惊讶地,化合物B也以浓度依赖的方式降低亚硝酸盐生成量,但效果不如化合物A。
在与下式:
的根据WO 2012/076696的姜黄素衍生物的对比测试中,还评价了化合物C降低亚硝酸盐水平的能力。
结果示于图3C中。
如这些结果所证实,化合物C比根据WO 2012/076696的姜黄素衍生物显著更有活性(几乎2倍),因为用2.5μM的化合物C和5μM的姜黄素衍生物观察到了相同的亚硝酸盐生成量的下降。
在与以上描述的对比化合物CORM-401和对比化合物J的对比测试中,还评价了化合物C降低亚硝酸盐水平的能力。
结果示于图3D中。
表1.对比化合物富马酸二甲酯、对比化合物J和化合物A的抗炎效力的比较。
在用0.5μg/ml脂多糖(LPS)±5μM的对比化合物富马酸二甲酯、对比化合物J或化合物A温育24h的细胞中测量亚硝酸盐水平。所有值(平均±SEM)均显示为单独LPS的百分比。*p<0.05对比于单独LPS;对比于富马酸二甲酯;对比于对比化合物。
如这些结果所证实,化合物C比富马酸衍生物(对比化合物J)显著更有活性(几乎5倍),因为用1μM的化合物C和5μM的富马酸衍生物J观察到了几乎相同的亚硝酸盐生成量的下降。此外,对比化合物CORM-401没有活性。
在亚硝酸盐生成量测试中,还将化合物A与富马酸二甲酯衍生物(对比化合物J)作对比。如表1所示,化合物A允许由LPS引起的炎症的显著降低(63.3%),其为由相同浓度的富马酸二甲酯衍生物(对比化合物J)所诱导的降低的3倍多。因此这些结果证明,富马酸部分与CORM的组合带来了疾病治疗中,更特别地炎症治疗中的很大好处。
在对比亚硝酸盐生成量测试中评估了下式:
(WO 2008/003953的化合物364)的根据WO 2008/003953的CORM衍生物。
如图3E所示,WO 2008/003953的化合物364在高达100μM的浓度能够降低亚硝酸盐生成量不到50%。因此,WO 2008/003953的化合物364远不如本发明的化合物A有活性,所述化合物A在低至5μM的浓度能够降低亚硝酸盐生成量超过60%(表1)。
实施例3.3:本发明化合物的细胞毒性的评价
化合物A和化合物B对RAW巨噬细胞的细胞毒性的评价
使用细胞毒性检测试剂盒(LDH)(Roche Applied Science)测量由受损细胞释放的乳酸脱氢酶,在用升高浓度的化合物A或化合物B温育24h后的BV2小胶质细胞中,和用下式:
的根据WO 2012/076696的姜黄素衍生物的RAW巨噬细胞中评估细胞毒性。使用在介质中制备的X-100Triton溶液(2%)作为阳性对照(100%细胞毒性)。根据制造商的说明进行测试。简言之,在温育结束时,将细胞板以300×g离心5min,将100μl不含细胞的上清液转移至96孔板中。向上清液中加入反应混合物,在温和振荡下将板室温避光温育10min。在485nm处测量吸光度。
数据表示为通过用2%的triton处理细胞而释放的LDH(100%毒性)的百分比。
结果示于图4中。
在低于50μM以下的浓度,化合物A的细胞毒性是不显著的。
化合物B仅在100μM的浓度显示出细胞毒性,且低于该浓度没有细胞毒性。
相比之下,根据WO 2012/076696的姜黄素衍生物在20μM的浓度已经对RAW巨噬细胞显示出细胞毒性。
化合物A对角化细胞的细胞毒性的评价
在用升高浓度的化合物A温育24h后的角化细胞中评估细胞毒性。
在补充有1%青霉素/链霉素、1%丙酮酸钠和10%胎牛血清的达尔伯克氏改良伊格尔培养基(DMEM)中培养人角化细胞(HaCaT细胞)。将聚集的细胞用PBS洗涤,并根据实验方案用不同浓度的化合物A处理不同的一段时间。以0.1%将化合物A溶解于DMSO中,作为完全培养基中的最终浓度。
当24孔板中的细胞聚集时,用新鲜介质替代培养基,然后用不同浓度(1-100μM)的化合物A处理24h。在37℃湿润的5%CO2中将细胞温育过夜。使用100%的DMSO作为阳性对照。24h后,用MTT溶液(0.5mg/ml)将细胞温育1h 30min。活细胞用MTT处理产生了深蓝色的甲臜产物,而在死细胞中观察不到染色。
结果示于图4E中。
在低于50μM的浓度,化合物A的细胞毒性不显著。
化合物A对THP-1细胞的细胞毒性的评价
在用升高浓度的化合物A温育24h后的THP-1细胞中评估细胞毒性。
在补充有1%青霉素/链霉素、1%丙酮酸钠和10%胎牛血清的RPMI1640-Glutamax培养基(RPMIc)中培养人单核细胞(THP-1细胞)。将聚集的细胞用PBS洗涤,并根据实验方案用不同浓度的化合物A处理不同的一段时间。以0.1%将化合物A溶解于DMSO中,作为完全培养基中的最终浓度。
在处理之前,将细胞用RPMI洗涤一次,计数,然后以1x106细胞/ml接种。然后用不同浓度(1-100μM)的化合物A将细胞处理24h。24h后,收集不同情况的上清液,使用细胞毒性检测试剂盒(LDH)(Roche,德国)根据制造商的说明进行LDH测试。使用2%的Triton作为阳性对照。
结果示于图4E中。
在低于100μM的浓度,化合物A的细胞毒性不显著。
实施例3.4:Nrf-2表达的评价
由化合物A、化合物B、化合物C诱导的Nrf-2表达的评价,以及化合物A和化合物C诱导的HO-1表达的评价。
将BV2小胶质细胞用20μM的化合物A或化合物B温育2h,以评价Nrf2的核移位。使用来自Active Motif(La Hulpe,比利时)的核提取试剂盒根据制造商的说明分离核部分(nuclear fraction),并储存在-80℃。使用Pierce BCA蛋白质测试试剂盒(Thermo Scientific)测量蛋白质浓度。
为了确定HO-1蛋白质表达,将BV2小胶质细胞用升高浓度的化合物A温育6h。在温育结束时,将细胞用冰冷的DPBS(-Ca,-Mg;Cell Culture,Life Technologies)洗涤,并在细胞裂解缓冲液(50mM HEPES,150mM NaCl,50mM NaF,50μM Na3VO4,1%v/v Triton X-100和1%哺乳动物蛋白酶抑制剂)中在4℃在30min的温育期间裂解。在4℃将裂解物以15,000×g离心10min;收集上清液并储存在-80℃。使用Pierce BCA蛋白质测试试剂盒(Thermo Scientific)测量蛋白质浓度。
将核提取物(50μg蛋白质/样品)和全细胞裂解物(20μg蛋白质/样品)分别再溶解(resolve)至10或12%丙烯酰胺凝胶上,并将蛋白质转移至聚偏二氟乙烯膜(Millipore,Brussels,比利时)上。在含有0.1%v/v吐温20和5%w/v脱脂奶粉的1×Tris-缓冲盐水(pH 7.5)中将膜在室温阻断1h,并在4℃用以下Nrf2的一级抗体:克隆C-20兔多克隆(Santa Cruz Biotechnology)温育过夜。然后,用偶联至辣根过氧化物酶的二级抗体(山羊抗小鼠或抗兔,Cell Signaling Technology,或者驴抗山羊,Jackson ImmunoResearch)将膜在室温温育1h。用化学发光底物(PierceThermo Scientific或Intense,Ozyme)探测条带,使用G:Box F3影像站和GeneSys软件(Syngene,剑桥,英国)拍摄图像。
结果示于图5A-C中。
BV2小胶质细胞暴露于20μM化合物A中持续2h强烈促进Nrf2在核中的积累。此外,在处理6h后,化合物A诱导了HO-1蛋白质表达的浓度依赖性增加。化合物B作为Nrf2活化剂和HO-1诱导剂均不太有效。
这些实验证明,在远低于毒性浓度范围的浓度,化合物A、化合物B和化合物C为HO-1的有效活化剂和经由Nrf2/HO-1轴(axis)的HO-1蛋白质表达的诱导剂。
此外,化合物C的活性与对比化合物CORM-401和J的比较显示,对于Nrf2和HO-1的活化,在相同浓度(5μm)时化合物C比富马酸衍生物(对比化合物J)和对比化合物CORM-401更有效(图5C)。
在角化细胞中化合物C对Nrf2和HO-1的诱导
角化细胞暴露于5μM化合物C中随着时间强烈促进Nrf2在核中的积累和HO-1蛋白质表达(图5D)。
这些实验证明在角化细胞中,在远低于毒性浓度范围的浓度,化合物C为经由Nrf2/HO-1轴的HO-1的有效活化剂。因此,其可被用于治疗炎性疾病,更特别地用于治疗皮肤炎症如伤口愈合或银屑病。
在THP-1中化合物C对Nrf2和HO-1的诱导
如对于角化细胞和BV2小胶质细胞,THP-1细胞暴露于化合物C中持续6小时强烈促进Nrf2在核中的积累和HO-1蛋白质表达(图5E)。此外,当与对比化合物J比较HO-1活化时,从5μM化合物C开始,化合物C诱导HO-1蛋白质表达的浓度依赖性增加,而用对比化合物J观察到相同的结果则需要10倍的该浓度。
这些实验证明在THP-1细胞中,化合物C也是经由Nrf2/HO-1轴的HO-1的有效活化剂。
因此,已证明本发明的化合物可被用于治疗炎性疾病。
在THP-1中化合物A、B和C对Nrf2和HO-1的诱导
将THP-1细胞暴露于不同浓度的化合物A、B和C中持续6小时。如上面用BV2小胶质细胞所示,化合物A和C诱导HO-1蛋白质表达的浓度依赖性增加和Nrf2活化,而化合物B作为Nrf2活化剂和HO-1诱导剂均不太有效(图5F)。
这些实验证明,仅包含一个CORM基团(也被称为单CORM,即式(I)的化合物,其中R1代表-Q’-Y)的本发明的化合物(如化合物A和化合物C)为有效的Nrf2活化剂和HO-1蛋白质表达诱导剂。
实施例4:在用LPS处理的人巨噬细胞中,本发明化合物对TNF-α生成量的评价
在补充有10%胎牛血清、1%丙酮酸钠、1%青霉素-链霉素和50μMβ-巯基乙醇的RPMI 1640GlutaMax(Gibco,法国)中,在37℃和5%CO2中保持人THP-1细胞(ATCC;#TIB-202)。将细胞接种至24孔板中(5×105/孔/ml),并用佛波脂(PMA(50ng/ml))处理24h以诱导巨噬细胞分化。然后,将人巨噬细胞用PBS洗涤,用介质温育,单独用LPS处理24h或者在各种化合物的存在下用LPS处理24h。24h后,收集培养基并储存在-20℃。根据制造商的说明(BD OptEIA Set)进行TNF-α的ELISA测试。简言之,将板(F96 maxisorp-Nunc-immuno,Thermofischer)用捕捉抗体包被过夜。然后,将板用PBS/吐温0.05%洗涤,并在37℃用PBS/FBS 10%使板饱和1h。洗涤几次后,将标准品和样品在室温温育2h。然后,将检测抗体和链霉亲和素-HRP在室温温育1h。最后,加入底物(TMB),导致蓝颜色。用硫酸使反应停止,导致黄颜色。使用酶标仪在450nm处读板。结果表示为皮克每ml。
如图7所示,人巨噬细胞暴露于LPS持续24h导致TNF-α生成量的显著增加,即炎症的标志。当用升高浓度(1-10μM)的化合物C、D或E共处理巨噬细胞时,这种作用显著减弱。化合物D看起来是三个受试化合物中最有效的。
实施例5:在用LPS处理的THP-1细胞中,本发明化合物对TNF-α生成量的评价
将如实施例3.3中所培养的人THP-1细胞单独用LPS(100μg/ml)处理24h,或者在5μM化合物C、化合物E或对比的富马酸二甲酯(DMF)的存在下用LPS(100μg/ml)处理24h。如图8所示,化合物C和化合物E均显著降低TNF-α生成量,而富马酸二甲酯(DMF)没有任何作用。
- 还没有人留言评论。精彩留言会获得点赞!