一种稳定表达抑癌基因REIC/Dkk3的人脐带间充质干细胞及其构建方法和应用与流程
本发明涉及生物医药技术领域,具体地说,是一种具备间充质干细胞特性,并被抑癌基因REIC/Dkk3修饰的人脐带间充质干细胞及其构建方法及其制药用途。
背景技术:
Wnt信号通路作为一种在进化中高度保守的信号通路,在生长、发育、代谢等多种生物学过程中发挥重要作用。Wnt通路的失控与癌症也密切相关,国内外众多研究证明,Wnt/β-catenin信号通路异常活化在膀胱肿瘤的发生发展中起着重要作用。REIC/Dkk-3是Wnt/β-catenin信号通路的抑制剂,国外已有研究证明,在膀胱肿瘤中,搭载REIC/Dkk-3基因的腺病毒能够显著地诱导肿瘤细胞凋亡,减缓肿瘤生长,并对多重耐药的肿瘤细胞也存在有效作用,这让我们看到了REIC/Dkk-3基因用于肿瘤靶向治疗的潜力。
随着对肿瘤发生发展分子机制认识的深入,人们已普遍意识到肿瘤是一种基因病,纠正发生缺陷的基因可为肿瘤的治疗提供新的希望。肿瘤基因治疗的临床研究开展已将近20年,基因治疗药物经历了从裸露DNA、非病毒载体基因药物到病毒类基因药物的发展过程。1995年之后,病毒载体由于转导效率较高且可用于体内和体外的基因转导,逐渐成为基因治疗研究和临床应用的主要工具,最主要的病毒载体有腺病毒及慢病毒,其中慢病毒载体较腺病毒载体相比,慢病毒载体具有可感染非分裂细胞、目的基因可整合至靶细胞基因组并长期表达且免疫反应小等优点,适于体内基因治疗,因此成为理想的基因转移载体。
综上可见搭载REIC/Dkk-3的慢病毒载体(Ad-REIC)有望成为体内基因治疗的有效载体,但该载体仍缺乏针对肿瘤细胞的靶向迁移能力和趋向性,而间充质干细胞(MSCs)不仅是基因的理想载体,而具有良好的肿瘤组织定向趋化能力,是肿瘤基因治疗的研究热点,因此借助基因工程技术构建REIC/Dkk3修饰的脐带间充质干细胞,以达到靶向治疗膀胱肿瘤的目的。
技术实现要素:
本发明的第一个目的是针对现有技术中的不足,提供一种表达抑癌基因REIC/Dkk-3的人脐带间充质干细胞的构建方法。
本发明的第二个目的是,提供如上所述表达抑癌基因REIC/Dkk-3的人脐带间充质干细胞的制药用途。
本发明的第三个目的是,提供一种用于靶向治疗膀胱肿瘤的药物组合物/试剂盒。
为实现上述第一个目的,本发明采取的技术方案是:
一种表达抑癌基因REIC/Dkk-3的人脐带间充质干细胞的构建方法,所述方法利用携带REIC/Dkk-3的慢病毒感染人脐带间充质干细胞,使人脐带间充质干细胞持续稳定表达REIC/Dkk-3,具体包括如下步骤:
(1)表达REIC/Dkk-3的慢病毒载体构建:根据GeneBank已有的REIC/Dkk-3的基因序列,设计扩增引物,提取总RNA,经聚合酶链式反应PCR合成得到REIC/Dkk-3的基因片段,酶切载体GV208,建立PCR产物线性表达载体,进一步转化抽提质粒,共转染包装细胞293T细胞,出现强绿色荧光并有细胞融合现象时,收集含慢病毒颗粒的细胞上清液,并测定病毒滴度;
(2)人脐带间充质干细胞的培养:取足月正常妊娠剖宫产健康胎儿的脐带,以PBS充分洗涤,剔除脐动、静脉,以α-MEM原代培养人脐带间充质干细胞,对分离培养的细胞进行表型鉴定,并置37℃、5%CO2,饱和湿度培养箱,待细胞80%融合时,胰酶消化1:3传代,连续培养至3代后采用流式细胞学技术检测细胞表面标志物;
(3)重组慢病毒转染人脐带间充质干细胞及鉴定:将携带REIC/Dkk-3基因的重组慢病毒转染入人脐带间充质干细胞,通过实时定量PCR及蛋白质印迹检测法检测REIC/Dkk-3表达情况,使人脐带间充质干细胞持续稳定表达REIC/Dkk-3,并用GFP标记。
进一步,所述引物序列如下:
上游引物为:5’-GTAAGTTTCCCCTCTGGCTTG-3’
下游引物为:5’-AAGCACCAGACTGTGAAGCCT-3’。
进一步,所述构建方法还包括如下步骤:
REIC/Dkk-3修饰的HUC-MSCs构建及鉴定:取对数生长期的P2代HUC-MSCs,Triple消化、离心后将细胞重选,调整细胞密度至约4×104/ml,培养12小时;待细胞贴壁完成后,观察细胞融合度,当细胞融合度达到约40%-50%后即可加入慢病毒溶液,保证转染3天后细胞贴满整个12孔板底部,以便观察转染效率;采用上述构建并浓缩及鉴定后的慢病毒溶液,调整复感染指数(MOI)控制在75;将间充质干细胞融合度达到40%-50%时的细胞培养基弃置,加入新鲜培养基300ul,取200ul新鲜培养基,加入适量病毒溶液,调节MOI至75-100后,均匀滴入12孔板各孔中,恒温摇床充分混匀,培养24小时后,将含有慢病毒的培养基弃置,更换新鲜培养基,继续培养2天后,观察细胞状态及荧光强度;以绿色荧光的表达水平作为慢病毒转染效率的考量指标。
进一步,所述构建方法还包括如下步骤:
(1)取足月正常妊娠剖宫产胎儿的脐带,浸入含抗生素的0.9%生理盐水中,4℃保存;
(2)在超净台内取出脐带,用生理盐水冲洗净脐动脉和脐静脉内的残余血液,用止血钳和剪刀剔除上述血管,将脐带剪成1mm3大小的组织块;
(3)加入质量/体积比为0.1%的II型胶原酶至完全覆盖组织块,置于培养箱内持续消化6h,100目筛网过滤收集细胞;
(4)生理盐水冲洗细胞3次,每次2000rpm,8min;
(5)用含10%FBS的DMEM/F12培养液重悬细胞,调整细胞密度为1x106cells/ml,接种于T25培养瓶中,置于37℃、饱和湿度、5%C02培养箱内培养;
(6)3天后全量换液,弃去未贴壁的细胞与杂质,以后根据细胞生长情况,观察致细胞80%融合时,胰酶消化1:3传代,连续培养至3代后采用流式细胞学技术再次检测细胞表面标志物。
进一步,所述脐带间充质干细胞有效表达REIC/Dkk-3,并对肿瘤存在趋向作用。
为实现上述第二个目的,本发明采取的技术方案是:
如上任一所述表达抑癌基因REIC/Dkk-3的人脐带间充质干细胞的制药用途,所述药物用于制备人膀胱肿瘤靶向治疗药物。
为实现上述第三个目的,本发明采取的技术方案是:
一种用于靶向治疗膀胱肿瘤的药物组合物,所述药物组合物包括如上所述表达抑癌基因REIC/Dkk-3的人脐带间充质干细胞。
一种用于靶向治疗膀胱肿瘤的试剂盒,所述试剂盒包括含有表达抑癌基因REIC/Dkk-3的表达载体、人脐带间充质干细胞或抗癌剂的前药。
本发明优点在于:
1、本发明制备的表达REIC/Dkk-3的人体脐带间充质干细胞既能拥有干细胞对肿瘤的靶向迁移功能,又很好的表达REIC/Dkk-3基因,以达到诱导肿瘤细胞凋亡,抑制肿瘤生长的作用,是对膀胱肿瘤靶向治疗极具潜力的治疗载体。
2、本发明根据GenBank中REIC/Dkk-3的基因序列,运用PCR技术进行体外扩增以得到目的基因片段,利用重组技术将编码人REIC/Dkk-3的基因片段转入慢病毒载体中,构建含人REIC/Dkk-3的重组慢病毒载体,然后将该慢病毒载体转染人脐带间充质干细胞。利用慢病毒载体在人脐带间充质干细胞内的复制,使人脐带间充质干细胞持续稳定表达REIC/Dkk-3。
附图说明
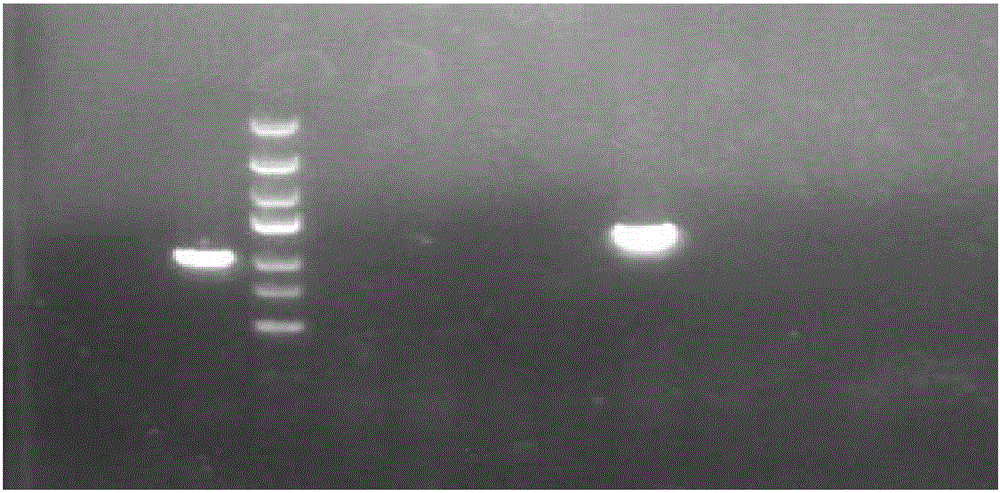
附图1为Dkk-3目的基因片段获取后电泳结果。
附图2为重组质粒构建PCR鉴定结果。
附图3为质粒转染293T细胞后目的蛋白表达WB鉴定。
附图4为质粒转染293T细胞后GFP荧光表达鉴定。
附图5为病毒浓缩后滴度鉴定结果。
附图6为转染MSCs后Dkk-3组与空载体对照组比较。
附图7为转染MSCs后Dkk-3表达量检测。
具体实施方式
下面结合具体实施方式,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1表达REIC/Dkk-3的慢病毒载体的构建
总RNA提取
运用Trizol法提取总RNA,具体步骤如下:
(1)样品处理:取100mg组织置1.5ml离心管中,加入1ml Trizol充分匀浆,室温静置5min。
(2)加入0.2ml氯仿,振荡15s,静置2min。
(3)4℃离心,12000g×15min,取上清。
(4)加入0.5ml异丙醇,将管中液体轻轻混匀,室温静置10min。
(5)4℃离心,12000g×10min,弃上清。
(6)加入1ml 75%乙醇,轻轻洗涤沉淀。4℃,7500g×5min,弃上清。
(7)晾干,加入适量的去离子水溶解(65℃促溶10-15min)。
逆转录合成cDNA,PCR扩增。
(1)反应体系:
总RNA 2ug
Oligo(dT)15 1μl
离心,70℃水浴10min后立即冰水浴,稍离心
(2)反应体系(总体系20μl):
引物设计,PCR扩增
根据Genbank中REIC/Dkk-3的基因序列,设计引物,合成目的基因片段。
上游引物为:5’-GTAAGTTTCCCCTCTGGCTTG-3’
下游引物为:5’-AAGCACCAGACTGTGAAGCCT-3’
反应体系:
酶切载体GV208
酶切反应温度:37℃
酶切反应时间:2h
酶切反应体系:
重组质粒构建
(1)PCR产物交换入线性表达载体得到pGC-LV
反应体系(μL):
制备感受态细胞
配置下述溶液:
(1)0.1M CaCl2溶液,以0.22μm过滤除菌。
(2)250mM KCl2溶液。
(3)2M MgCl2溶液,高压灭菌。
(4)SOB:1ml 250mM KCl2溶液加入100ml LB,以5M NaOH调PH值至7.0,高压灭菌,临用前加入0.5ml 2M MgCl2溶液。
用氯化钙制备新鲜的大肠杆菌感受态细胞
(1)从于37℃培养16小时的新鲜平板中挑取一个单菌落,转到一个含有100ml LB培养基的1L烧瓶中,于37℃剧烈振摇培养3小时(旋转摇床,300转/分)。
(2)在无菌条件下将细菌转移到一个无菌、一次性使用的、用冰预冷的50ml聚丙烯管中,在冰上放置10分钟,使培养物冷却至0℃
(3)于4℃以4000转/分离心10分钟,回收细胞。
(4)倒出培养液,将管倒置1分钟,使最后残留的痕量培养液流尽。
(5)以10ml用冰预冷的0.1mol/L CaCl2重悬每份沉淀,放置于冰浴上。
(6)于4℃,以4000转/分离心10分钟,回收细胞。
(7)倒出培养液,将管倒置1分钟,使最后残留的痕量培养液流尽。
(8)每50ml初始培养物用2ml用冰预冷的0.1mol/LCaCl2重悬每份细胞沉淀。
(9)将细胞分装成小份,放于-70℃冻存。
转化
(1)用冷却的无菌吸头从每种感受态细胞悬液中各取200μL转移到无菌的微量离心管中,每管加10μL连接液,轻轻旋转以混匀内容物,在冰中放置30分钟。
(2)将管放到预加温到42℃的循环水浴中放好的EP管架上,恰恰放置90秒,不要摇动EP管架。
(3)快速将管转移到冰浴中,使细胞冷却l-2分钟。
(4)每管加800μL LB培养基。用水浴将培养基加温至37℃,然后将管转移到37℃摇床上,温育45分钟使细菌复苏。
(5)将150μL已转化的感受态细胞转移到AMP抗性(100ug/ml)的LB琼脂培养基上。
(6)将平板置于室温直至液体被吸收。
(7)倒置平皿,于37℃培养,16小时。
抽提pGC-LV质粒DNA,PCR扩增测序
挑选没有突变的菌落,抽提质粒。按照质粒抽提试剂盒说明书操作,实验步骤如下:
(1)将培养过夜的2ml菌液移至2ml离心管中,最高速离心30sec,弃尽上清;
(2)在细菌沉淀中加入250μl Solution1液,振荡至彻底悬浮;
(3)加入250μl Solution 2溶液,立即温和地上下翻转离心管5-10次以混匀,使菌体充分裂解直至形成透亮的溶液,此步骤应在5min内完成;
(4)加入400μl 4℃预冷的Solution 3溶液,立即温和并充分地翻转离心管5-10次以混匀,室温静置2min,最高速离心10min;
(5)将上清倒入新的离心管中,再次最高速离心10min;
(6)小心吸取上清转移到DNA吸附柱中(吸附柱置于废液收集管中),离心1min,弃滤液,并将吸附柱置于同一收集管中;
(7)在吸附柱中加入500μlW1液,离心30sec,弃滤液,并将吸附柱置于同一收集管中;
(8)在吸附柱中加入700μl W2液,离心30sec,弃滤液,并将吸附柱置于同一收集管中,重复一次;
(9)最高速离心1min;
(10)将吸附柱移入新的干净的1.5ml离心管中,在DNA吸附膜中央加入60μl预热到60℃的去离子水,室温静置1min,最高速离心1min,得到的即为质粒DNA溶液。PCR测序,反应条件如前,测序结果见图,通过GENBANK对照,碱基序列与REIC/Dkk-3基因完全一致。表明PCR扩增得到的目的基因完全正确。
慢病毒转染293T细胞及包装
(1)转染前24h,用胰蛋白酶消化对数生长期的293T细胞,以含10%血清的培养基调整细胞密度为1.2×107细胞/20ml,重新接种于15cm细胞培养皿,37℃、5%CO2培养箱内培养。24h待细胞密度达70%-80%时即可用于转染。
(2)转染前2h将细胞培养基更换为无血清培养基。
(3)向一灭菌离心管中加入所制备的各DNA溶液(pGC-LV载体20μg,pHelper 1.0载体15μg,pHelper 2.0载体10μg),与相应体积的Opti-MEM混合均匀,调整总体积为2.5ml,在室温下温育5分钟。
(4)将Lipofectamine 2000试剂轻柔摇匀,取100μl Lipofectamine 2000试剂在另一管中与2.4ml Opti-MEM混合,在室温下温育5分钟。
(5)把稀释后的DNA与稀释后的Lipofectamine 2000进行混合,轻轻地颠倒混匀,不要振荡。必须在5分钟之内混合。
(6)混合后,在室温下温育20分钟,以便形成DNA与Lipofectamine 2000稀释液的转染复合物。
(7)将DNA与Lipofectamine 2000混合液转移至293T细胞的培养液中,混匀,于37℃,5%CO2细胞培养箱中培养。
(8)培养8h后倒去含有转染混和物的培养基,每瓶细胞加入20ml的PBS液,轻轻左右晃动一下培养瓶以洗涤残余的转染混和物,然后倒去。
(9)每瓶细胞中加入含10%血清的细胞培养基25ml,于37℃、5%CO2培养箱内继续培养48小时。
慢病毒的收获及浓缩
(1)收集转染后48小时(转染即可为0小时计起)的293T细胞上清液。
(2)于4℃,4000g离心10min,除去细胞碎片。
(3)以0.45μm滤器过滤上清液于40ml超速离心管中。
(4)把病毒粗提液样品加入到过滤杯中(最多19ml),盖上盖子。将过滤杯插到滤过液收集管中。
(5)组合好后,做好平衡,放在转头上。
(6)在4000×g离心,至需要的病毒浓缩体积。通常需要的时间为10-15分钟。
(7)离心结束后,取出离心装置,将过滤杯和下面的滤过液收集杯分开。
(8)将过滤杯倒扣在样品收集杯上。
(9)离心力不超过1000g,时间为2分钟。过高转速会导致样品损失。把过滤杯从样品收集杯上移开。样品收集杯中的即为病毒浓缩液。
(10)将病毒浓缩液移出,分装后保存在病毒管中,-80度长期保存。取其中一支进行病毒生物学滴度测定。
病毒滴度测定
荧光法测定滴度,步骤如下
(1)测定前一天,为测定滴度所需的293T细胞铺板,96孔板,每个孔加4×104个细胞,体积为100μl。
(2)根据病毒的预期滴度,准备7~10个无菌的Ep管。在每个管中加入90μl的无血清培养基。
(3)取待测定的病毒原液10μl加入到第一个管中,混匀后,取10μl加入到第二个管中。继续相同的操作直到最后一管。
(4)选取所需的细胞孔,吸去90μl培养基,丢弃。加入90μl稀释好的病毒溶液。放入培养箱培养。
(5)24小时后,加入完全培养基100μl。
(6)4天后,观察荧光表达情况。荧光细胞数随稀释倍数的增加而减少。
(7)根据荧光结果,计算病毒滴度。
实施例2人脐带间充质干细胞的分离培养及鉴定
(1)经父母授权同意,取足月正常妊娠剖宫产胎儿的脐带,浸入含抗生素的0.9%生理盐水中,4℃保存;
(2)在超净台内取出脐带,用生理盐水冲洗净脐动脉和脐静脉内的残余血液,用止血钳和剪刀剔除上述血管,将脐带剪成1mm3大小的组织块;
(3)加入质量/体积比为0.1%的II型胶原酶至完全覆盖组织块,置于培养箱内持续消化6h,100目筛网过滤收集细胞。
(4)生理盐水冲洗细胞3次,每次2000rpm,8min;
(5)用含10%FBS的DMEM/F12培养液重悬细胞,调整细胞密度为1x106cells/ml,接种于T25培养瓶中,置于37℃、饱和湿度5%C02培养箱内培养;
(6)3天后全量换液,弃去未贴壁的细胞与杂质,以后根据细胞生长情况,观察致细胞80%融合时,胰酶消化1:3传代,连续培养至3代后采用流式细胞学技术再次检测细胞表面标志物,MSCs为高表达简称值细胞表面抗原CD44,CD90,CD105,低表达或者不表达造血前提细胞标志抗原CD34、白细胞标志抗原CD45、淋巴细胞表面抗原CD19、单核细胞/巨噬细胞表面抗原CD14。
实施例3重组慢病毒转染人脐带间充质干细胞及功能鉴定
(1)准备细胞:将培养的第三代人脐带间充质干细胞转至24孔板内,每个孔内细胞数在3~5×104左右,铺板时细胞的融合率为50%左右,每孔培养基体积为100μl;进行病毒感染时细胞的融合度需为70%左右。
(2)将准备好的重组慢病毒从-80℃中取出,置于冰上融化。
(3)使用移液器吸取准确体积的病毒液加入准备好的培养基,吸去原细胞培养器皿中的培养基,并在目的细胞和对照细胞中分别加入计算好的病毒液混匀后,放于二氧化碳培养箱(37℃、5%CO2)孵育过夜。
(4)感染后观察细胞状态,及时更换培养基,一般在24小时候后将含有慢病毒的培养液更换成正常培养液。
(5)第六天,感染效率检测:在倒置荧光显微镜观察荧光,估计慢病毒感染目的细胞的效率。
(6)持续按照已述方法培养干细胞,western blotting鉴定,使其稳定表达REIC/Dkk-3后用GFP标记。
Western blotting检测REIC/Dkk-3表达
(1)常规蛋白电泳分离样品;
(2)PVDF膜用甲醇浸湿,去离子水冲洗,再转移到Transferring buffer中平衡20min,电泳胶置Transferringbuffer中平衡20min,裁4张与胶同样大小的Whatman3mm滤纸,置Transferringbuffer中平衡;
(3)按下列顺序装好blot:负极—2层滤纸—电泳胶—PVDF膜—2层滤纸—正极,在这一步,要注意PAGE胶上的预染Marker是否清楚,为了避免在转膜之后膜上的Marker不清楚,可以在这一步,当胶和膜叠在一起时,对应胶上的Marker,用圆珠笔在膜的相应位置做上记号;
(4)5%脱脂牛奶/PBST 20ml,水平摇床,室温封闭1h;也可以先室温封闭若干时间,4℃过夜;
(5)用PBST稀释一抗,5%BSA,稀释倍数:1000倍,将膜封在塑料袋中,不留气泡,用胶带固定在可垂直旋转的小摇床上,偏转角度70-80度,水平摇床,室温1hr;先用PBST短暂地洗2次,再用>4ml/cm2(膜面积)的PBST,室温洗15min;
(6)用PBST稀释二抗,5000倍或10000倍稀释5%BSA,水平摇床,室温1hr;先用PBST短暂地洗2次,再用>4ml/cm2(膜面积)的PBST,室温洗15min;
(7)将检测试剂(ECLplus)从4℃取出,在打开前平衡至室温,检测试剂的A液与B液以40:1的比例混合,检测试剂的用量为0.1ml/cm2;
(8)膜置于吸水纸上干燥,蛋白面朝上,检测试剂加在膜上,覆盖整个膜表面,室温2min;
(9)用镊子夹起PVDF膜,使膜的下边角搭在纸巾上,吸干多余的检测试剂;
(10)PVDF膜用保鲜膜封起,暗室曝光,时间10sec。
REIC/Dkk-3修饰的HUC-MSCs构建及鉴定
第一天,取对数生长期的P2代HUC-MSCs,Triple消化、离心后将细胞重选,调整细胞密度至约4×104/ml,培养12小时。待细胞贴壁完成后,观察细胞融合度,当细胞融合度达到约40%-50%后即可加入慢病毒溶液,保证转染3天后细胞贴满整个12孔板底部,以便观察转染效率。采用上述构建并浓缩及鉴定后的慢病毒溶液,以不同感染复数(multiple of infection,MOI,分别为50、60、75、80、90、100)的重组慢病毒感染hUC-MSCs,通过EGFP的表达筛选最佳MOI。重组慢病毒载体转染hUC-MSCs后96h荧光倒置显微镜观察绿色荧光的表达情况。MOI值为75感染效率为80%以上,而75以上MOI值感染效率无明显升高,MOI=90时感染效率反而下降,故最佳MOI值为75。选择MOI控制在75-100之间。将间充质干细胞融合度达到40%-50%时的细胞培养基弃置,加入新鲜培养基300ul,取200ul新鲜培养基,加入适量病毒溶液,调节MOI至75-100后,均匀滴入12孔板各孔中,恒温摇床充分混匀,培养24小时后,将含有慢病毒的培养基弃置,更换新鲜培养基,继续培养2天后,观察细胞状态及荧光强度。以绿色荧光的表达水平作为慢病毒转染效率的考量指标。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!