一种氟康唑的制备方法与流程
本发明涉及氟康唑
技术领域:
,尤其涉及一种氟康唑的制备方法。
背景技术:
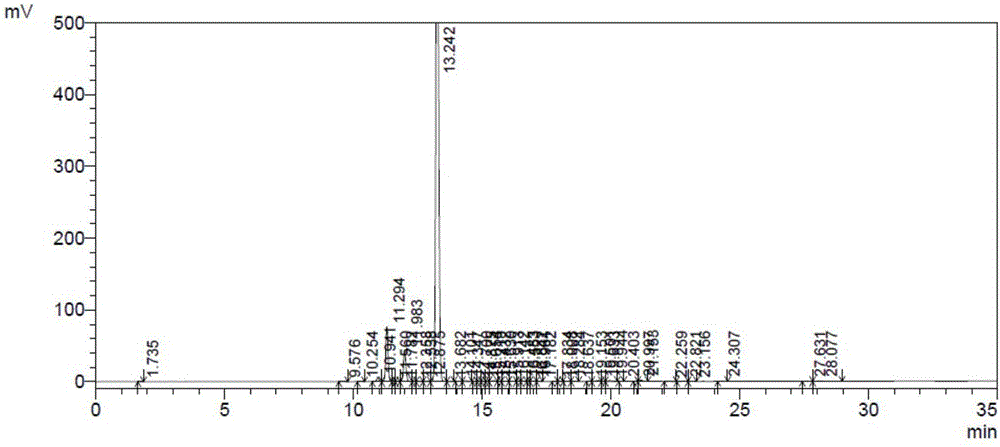
:氟康唑,商品名为大扶康,为新型三氮唑类抗真菌药,其结构式如下:氟康唑是上世纪八十年代美国辉瑞(Pfizer)公司研制的广谱、高效的抗真菌药物,1992年国内开始生产并应用于临床治疗。氟康唑主要通过乙酯真菌细胞膜麦角甾醇的生物合成过程中的限速酶CYP51的生成来达到较强的抑菌效果(尤其对深部真菌),并且具有良好的低毒性,耐受性,高口服生物利用度和代谢稳定性。在治疗曲霉菌、全身念珠菌以及脑膜炎隐球菌感染方面应用广泛。目前,氟康唑的合成方法主要有:第一种,化合物I在碱存在下,与三甲基碘化亚砜发生Corey-Chaykovsky反应,得到环氧化物,环氧化物的甲磺酸盐在碱的存在下,被1,2,4-三氮唑开环得到氟康唑。这种Corey-Chaykovsky反应得到的产物收率不高。第二种,文献(ES8604939,ES8604940)报道,化合物I或者2,4-二氟苯甲酸酯与格氏试剂,与1-(1,2,4-三氮唑-1-基)-甲基卤代镁反应得到氟康唑,该反应的收率也较低;而且涉及到的格氏试剂的合成,较为复杂。第三种,文献(ES8604938,CN201010246142,CN201510061034)报道,以2,4-二氟溴苯为原料,制成格氏试剂2,4-二氟苯基卤代镁,然后与相应的酮反应,得到氟康唑,收率较低;其中,1,3-二氯(溴)丙酮腐蚀性强,稳定性差;1,3-双(1,2,4-三氮唑-1-基)丙酮的合成繁琐,且收率不高。可以看出,现有技术提供的氟康唑的制备方法收率较低,如何提高氟康唑制备收率是本领域目前急需解决的问题。技术实现要素:有鉴于此,本发明的目的在于提供一种氟康唑的制备方法,本发明提供的方法制备氟康唑的纯度和收率均较高。本发明提供了一种氟康唑的制备方法,包括:在碱催化剂的作用下,将式I结构化合物、三甲基溴化亚砜和1,2,4-三氮唑在溶剂中进行反应,得到氟康唑;优选的,所述碱催化剂为碱金属氢氧化物。优选的,所述溶剂为醇或含水醇溶液。优选的,所述反应的温度为50~80℃。优选的,所述反应的时间为2~12小时。优选的,所述式I结构化合物、三甲基溴化亚砜和1,2,4-三氮唑的摩尔比为1:(1~1.5):(1~1.5);所述式I结构化合物和碱催化剂的摩尔比为(2~4):1。优选的,所述反应完成后还包括:将得到的反应产物采用酸溶碱析法提纯,得到氟康唑。优选的,所述酸溶碱析法采用的酸为盐酸、硫酸、磷酸、甲磺酸或醋酸。优选的,所述酸溶碱析法采用的碱为氨水或碱金属氢氧化物。优选的,对反应产物采用酸溶碱析法提纯后还包括:将得到的提纯物采用异丙醇进行重结晶,得到氟康唑。与现有技术相比,本发明采用三甲基溴化亚砜制备氟康唑,三甲基溴化亚砜中溴离子的亲核性较弱,能够有效避免副反应的发生,从而提高氟康唑的产率和纯度。另外,采用三甲基溴化亚砜进行反应其摩尔质量利用率高,价格低廉,具有明显的成本优势。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。图1为本发明实施例提供的氟康唑的合成路线图;图2为本发明实施例2制备得到的氟康唑粗品高效液相色谱图;图3为氟康唑标准液相色谱图;图4为本发明实施例4制备得到的氟康唑高效液相色谱图。具体实施方式下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明提供了一种氟康唑的制备方法,包括:在碱催化剂的作用下,将式I结构化合物、三甲基溴化亚砜和1,2,4-三氮唑在溶剂中进行反应,得到氟康唑;在本发明中,所述反应的温度优选为50~80℃,更优选为55~70℃,最优选为60~65℃。在本发明中,所述反应的时间优选为2~12小时,更优选为3~7小时,最优选为4小时。在本发明中,所述碱催化剂优选为碱金属氢氧化物,更优选为氢氧化钾或氢氧化钠,最优选为氢氧化钾。本发明对所述式I结构化合物的来源没有特殊的限制,采用本领域技术人员熟知的式I结构化合物即可,可由市场购买获得,也可参照专利US5710280,按照下述步骤制备得到:(A)在三氯化铝的作用下,将间二氟苯和氯乙酰氯进行反应,得到2’,4’-二氟-α-氯乙酰苯;(B)将2’,4’-二氟-α-氯乙酰苯和4-氨基-1,2,4-三氮唑在溶剂中进行反应,得到中间产物;(C)在盐酸的作用下,将中间产物和亚硝酸钠进行反应,得到式I结构化合物。在本发明中,所述步骤(A)中的反应优选在氮气的保护下进行。在本发明中,所述步骤(A)中的反应温度优选为45~50℃。在本发明中,所述步骤(A)中的反应时间优选为1.5~2.5小时,更优选为2小时。在本发明中,所述步骤(A)优选为先将三氯化铝和间二氟苯混合,然后向得到的混合物中滴加氯乙酰氯。在本发明中,所述滴加氯乙酰氯的温度优选为10~15℃。在本发明中,所述三氯化铝、间二氟苯和氯乙酰氯的摩尔比优选为(1~1.2):1:(1~1.1),更优选为1.14:1:1.01。在本发明中,所述步骤(A)中的反应完成后,本发明优选将得到的反应产物溶解后萃取,然后进行洗涤旋干,得到2’,4’-二氟-α-氯乙酰苯。在本发明中,所述溶解的溶剂优选为二氯甲烷和水。在本发明中,所述萃取的试剂优选为二氯甲烷。在本发明中,所述旋干的温度优选为35~45℃,更优选为40℃。在本发明中,所述步骤(B)中反应的温度优选为80~90℃,更优选为85℃。在本发明中,所述步骤(B)中的反应时间优选为16~20小时,更优选为18小时。在本发明中,所述步骤(B)中的反应优选在搅拌的条件下进行。在本发明中,所述步骤(B)中的反应完成后,本发明优选将得到的反应产物冷却后抽滤,将得到的固体产物洗涤、抽滤、旋干,得到中间产物。在本发明中,所述冷却的温度优选为5~10℃。在本发明中,所述洗涤的试剂优选为二氯甲烷。在本发明中,所述旋干的温度优选为55~65℃,更优选为60℃。在本发明中,所述步骤(C)中的反应温度优选为35~40℃。在本发明中,所述步骤(C)中的反应时间优选为1~2小时。在本发明中,所述步骤(C)优选为将所述中间产物采用水溶解冷却至10℃以下,向其中加入盐酸,冷却至5℃以下,向其中加入亚硝酸钠溶液进行反应,得到式I结构化合物。在本发明中,所述亚硝酸钠溶液优选为亚硝酸钠水溶液。在本发明中,所述亚硝酸钠溶液的质量浓度优选为20~30%,更优选为25%。在本发明中,所述步骤(C)中的反应完成后,本发明优选将得到的反应产物冷却后调节体系的pH至中性,然后升温至室温进行抽滤和洗涤,将得到的洗涤物再次进行抽滤和旋干,得到式I结构化合物。在本发明中,将所述反应产物冷却的温度优选为20~30℃。在本发明中,优选采用氨水调节体系pH至中性。在本发明中,所述洗涤的试剂优选为水。在本发明中,所述旋干的温度优选为65~75℃,更优选为70℃。本发明对所述三甲基溴化亚砜和1,2,4-三氮唑的来源没有特殊的限制,可由市场购买获得。在本发明中,所述溶剂优选为醇溶液,更优选为醇的水溶液。在本发明中,所述醇溶液中的醇优选为甲醇、乙醇或异丙醇,更优选为异丙醇。在本发明中,所述式I结构化合物、三甲基溴化亚砜和1,2,4-三氮唑的摩尔比优选为1:(1~1.5):(1~1.5),更优选为1:(1~1.1):(1~1.3),最优选为1:1.05:1.2。在本发明中,式I结构化合物和碱催化剂的摩尔比优选为(2~4):1,更优选为(2.5~3):1,最优选为2.8:1。在本发明中,所述反应完成后,本发明优选将得到的反应液pH值调至中性,浓缩去除溶剂后萃取,再次将pH值调节至中性后过滤,得到氟康唑粗品。在本发明中,将反应液pH值调节至中性的试剂优选为盐酸,更优选为质量浓度为10%的盐酸溶液。在本发明中,所述萃取的试剂优选为乙酸乙酯,萃取后乙酸乙酯层采用酸溶液进行萃取,水层的pH值采用碱调制7~8,将析出的固体抽滤,得到氟康唑粗品。在本发明中,所述酸溶液优选为盐酸溶液,更优选为质量浓度为5%的盐酸溶液。在本发明中,所述氟康唑粗品的纯度为85~90%,收率可达到80%。得到氟康唑粗品后,本发明优选采用酸溶碱析法对氟康唑粗品提纯后采用异丙醇重结晶,得到氟康唑纯品。在本发明中,将所述氟康唑粗品进行酸溶碱洗析之前优选对氟康唑粗品进行水洗。在本发明中,所述水洗过程中,氟康唑粗品和水的质量比优选为1:(3~10),更优选为1:(4~6),最优选为1:5。本发明将氟康唑粗品水洗加热溶清后冷却至室温(20~25℃),无需干燥,可直接进行酸溶碱析提纯。本发明对所述酸溶碱析的方法没有特殊的限制,采用本领域技术人员熟知的酸溶碱析的技术方案即可,可以按照下述方法进行:将氟康唑粗品溶于酸中,加入脱色剂进行脱色过滤,将得到的滤液采用碱调制pH值至7~8,析出大量白色固体,即为氟康唑。在本发明中,所述酸优选为酸溶液,所述酸溶液的质量浓度优选为15~25%,更优选为20%。在本发明中,所述酸优选为盐酸、硫酸、磷酸、甲磺酸或醋酸。在本发明中,所述脱色剂优选为活性炭。在本发明中,所述碱优选为氨水、氢氧化钠或氢氧化钾。将所述氟康唑粗品进行酸溶碱析后,本发明优选将得到的产物采用异丙醇进行重结晶,得到氟康唑纯品。本发明将所述产物进行重结晶之前优选将其冷却至室温(20~25℃)后过滤,将得到的滤饼干燥。在本发明中,所述过滤的方法优选为真空过滤。在本发明中,所述干燥的方法优选为真空干燥。在本发明中,所述干燥的温度优选为70~90℃,更优选为75~85℃,最优选为80℃。在本发明中,所述产物和异丙醇的体积/重量比优选为(2~10):1,更优选为(3~5):1。本发明对所述重结晶的方法没有特殊的限制,采用本领域技术人员熟知的重结晶的技术方案进行即可,可将得到的产物在异丙醇中加热溶解后冷却至室温,过滤后进行干燥,得到氟康唑纯品。在本发明中,所述过滤和干燥的方法与上述技术方案所述过滤和干燥的方法一致,在此不再赘述。本发明将氟康唑粗品进行提纯后,纯度可达99%以上,收率可达75%。本发明提供的方法采用三甲基溴化亚砜制备氟康唑,有效利用的摩尔质量高;一锅法合成氟康唑,成本降低,反应条件温和,易于操作,产品总收率达到60%,纯度可达99.0%以上,适合工业化生产。本发明以下实施例所用到的原料均为市售商品,式I结构化合物按照下述方法制备得到:氮气保护下,向2000mL反应瓶中加入305.5g无水三氯化铝(粉状,1.14mol)和228.8g间二氟苯(1.00mol),搅拌冷却至12.5℃,在10~15℃滴加228.6g氯乙酰氯(1.01mol),三氯化铝固体逐渐溶解,产生的酸气用碱液吸收。滴加完毕(2~3小时),撤去冷却,升温至45~50℃,在该温度保温反应2小时,冷却降温至15℃以下,加入686mL二氯甲烷搅拌稀释,滴加915mL水(注意:滴加过程中开始会析出类白色粘性物,约滴加到1/3时量最多,小心搅拌,后随水的加入粘性物逐渐溶解),控温室温以下。滴加完毕(1.5小时),在室温下搅拌,静置分液,水相用230mL二氯甲烷萃取,合并有机相,先用水230mL(1v/w)×5洗涤,使体系pH至中性。二氯甲烷层在40℃下旋蒸至干,得2’,4’-二氟-α-氯乙酰苯固体366.7g,纯度:99.56%,收率:95.94%。向2000mL反应瓶中加入191.3g4-氨基-1,2,4-三氮唑(1.20mol),2’,4’-二氟-α-氯乙酰苯的异丙醇溶液(固体361.1g,加685mL异丙醇溶解)和400mL异丙醇,搅拌加热升温至回流,在80~90℃之间反应约16~20小时,冷却降温至5~10℃,保温搅拌2小时,抽滤,固体用722mL二氯甲烷(2v/w)室温下搅拌洗涤,抽滤,固体在60℃下旋蒸干燥,得中间产物484.1g,收率:93.01%。向2000mL反应瓶中加入120.5g中间产物和458mL水,搅拌溶解冷却降温至10℃以下,加入91.5ml浓盐酸(2.50mol),继续冷却至5℃以下,滴加25%亚硝酸钠溶液(31.6g亚硝酸钠,1.04mol+94.8g水)控温-5~5℃,加完保温约半小时,自然升温至室温,加热至35~40℃,搅拌1~2小时,搅拌冷却至20~30℃,滴加氨水溶液调节体系pH至中性,升至室温,抽滤,固体用水约240mL室温下搅拌洗涤,抽滤,固体在70℃下旋蒸干燥得化合物I固体88.6g,纯度:99.56%,收率:90.49%。实施例1按照图1所示的合成路线制备氟康唑,图1为本发明实施例提供的氟康唑的合成路线图:1000mL反应瓶中,加入化合物I85.9g(1.0mol),三甲基溴化亚砜70.0g(1.05mol),1,2,4-三氮唑31.9g(1.20mol),搅拌下加入氢氧化钾溶液KOH(2.82mol)(60.9gKOH溶于345mL水)和345mL异丙醇,加热升温至60℃,在60~65℃保温3小时,HPLC检测原料小于1%,停止加热,在50℃下旋蒸除去异丙醇,向剩余产物中加入345mL水后冷却降温至15℃以下,滴加10%HCl溶液调节体系pH至中性,水相析出固体,用乙酸乙酯688mL溶解萃取,固体未全溶,分液,水相用乙酸乙酯172mL×6萃取,合并乙酸乙酯相,乙酸乙酯相用5%HCl溶液260mL×2萃取,合并HCl溶液,用乙酸乙酯172mL×2洗涤,再用甲苯172mL洗涤,加4.3g活性炭搅拌,垫硅藻土层抽滤,水洗,滤液冷却降温在10℃以下,加氨水调节pH至中性,保温搅拌析晶,抽滤,固体用170mL水搅拌打浆抽滤,得类白色固体,真空加热(80℃)干燥,得氟康唑粗品,类白色固体82.5g,纯度89.8%,收率80%。实施例21000mL反应瓶中,加入化合物I85.9g(1.0mol),三甲基溴化亚砜70.0g(1.05mol),1,2,4-三氮唑31.9g(1.20mol),搅拌下加入氢氧化钾溶液KOH(2.82mol)(60.9gKOH溶于627mL水)和63mL异丙醇,加热升温至60℃,在60~65℃保温3小时,HPLC检测原料小于1%,停止加热,在50℃下旋蒸除去异丙醇,向剩余产物中加入345mL水后冷却降温至15℃以下,滴加10%HCl溶液调节体系pH至中性,水相有固体析出,用乙酸乙酯688mL溶解萃取,固体未全溶,分液,水相用乙酸乙酯172mL×6萃取,合并乙酸乙酯相,乙酸乙酯相用5%HCl溶液260mL×2萃取,合并HCl溶液,用乙酸乙酯172mL×2洗涤,再用甲苯172mL洗涤,加4.3g活性炭搅拌,垫硅藻土层抽滤,水洗,滤液冷却降温在10℃以下,加氨水调节pH至中性,保温搅拌析晶,抽滤,固体用170mL水搅拌打浆抽滤,得类白色固体,真空加热(80℃)干燥,得氟康唑粗品,类白色固体82.5g,纯度86.3%,收率70%。对本发明实施例2制备得到的氟康唑粗品进行高效液相色谱检测,检测结果如图2所示,图2为本发明实施例2制备得到的氟康唑粗品高效液相色谱图,数据如表1所示,表1为本发明实施例2制备的氟康唑粗品的高效液相色谱数据;并与氟康唑标准液相色谱进行比对,如图3所示,图3为氟康唑标准液相色谱图,可以看出,本发明实施例提供的方法制备得到的目标产物氟康唑。表1本发明实施例2制备的氟康唑粗品的高效液相色谱数据实施例31000mL反应瓶中,加入化合物I85.9g(1.0mol),三甲基溴化亚砜70.0g(1.05mol),1,2,4-三氮唑31.9g(1.20mol),搅拌下加入氢氧化钾溶液KOH(2.82mol)(60.9gKOH溶于345mL水)和345mL异丙醇,加热升温至50℃,在50~55℃保温10小时,HPLC检测原料小于1%,停止加热,在50℃下旋蒸除去异丙醇,向剩余产物中加入345mL水后冷却降温至15℃以下,滴加10%HCl溶液调节体系pH至中性,水相有固体析出,用乙酸乙酯688mL溶解萃取,固体未全溶,分液,水相用乙酸乙酯172mL×6萃取,合并乙酸乙酯相,乙酸乙酯相用5%HCl溶液260mL×2萃取,合并HCl溶液,用乙酸乙酯172mL×2洗涤,再用甲苯172mL洗涤,加4.3g活性炭搅拌,垫硅藻土层抽滤,水洗,滤液冷却降温在10℃以下,加氨水调节pH至中性,保温搅拌析晶,抽滤,固体用170mL水搅拌打浆抽滤,得类白色固体,真空加热(80℃)干燥,得氟康唑粗品,类白色固体82.5g,纯度85.1%,收率72%。实施例4500mL四颈瓶中,加入实施例2制备得到的氟康唑粗品60.0g,纯化水300mL,加热至90℃溶清,2小时降至室温(20~25℃),在该温度保温2小时,抽滤得到白色固体;该固体无需干燥,直接进行酸溶碱析纯化:500mL四颈瓶中,加入该固体,水150mL,浓盐酸30mL,甲苯30mL,室温下搅拌半小时,分出甲苯层,水层用甲苯30mL×2洗涤,水层转至500mL四颈瓶中,加入活性炭5.0g,搅拌升温至50~60℃,1小时滴入浓氨水45mL,滴毕,pH为7~8,升温至90℃,固体溶清,2小时降至室温,在室温搅拌2小时,抽滤,用水25mL×2洗涤,得到白色固体在80℃真空干燥4小时以上,得到固体51.2g。该固体置于500mL四颈瓶中,加入异丙醇154mL,加热至溶清,2小时降温至室温,在室温搅拌2小时,抽滤,用异丙醇25mL×2洗,得到固体,在80℃真空干燥4~8小时,得到氟康唑45.0g,精制总收率:75%;纯度:99.52%,水分:0.21%。对本发明实施例4制备得到的氟康唑进行高效液相色谱检测,检测结果如图4所示,图4为本发明实施例4制备得到的氟康唑高效液相色谱图,数据如表2所示,表2为本发明实施例4制备的氟康唑的高效液相色谱数据。表2本发明实施例4制备的氟康唑的高效液相色谱数据PeakRet.TimeAreaHeightArea%110.80634026080.036211.1541745128960.186313.0969359597124778799.512413.53353602950.057515.31121922580.023615.89817282550.018716.689400640.004816.85459718510.063917.08011741900.0121019.0387241100.0081119.55011121650.0121219.96517312600.0181320.87116322540.0171420.99129824180.032Total94054551254411100.000由以上实施例可知,本发明提供了一种氟康唑的制备方法,包括:在碱催化剂的作用下,将式I结构化合物、三甲基溴化亚砜和1,2,4-三氮唑在溶剂中进行反应,得到氟康唑。与现有技术相比,本发明采用三甲基溴化亚砜制备氟康唑,三甲基溴化亚砜中溴离子的亲核性较弱,能够有效避免副反应的发生,从而提高氟康唑的产率和纯度。另外,采用三甲基溴化亚砜进行反应其摩尔质量利用率高,价格低廉,具有明显的成本优势。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1