一种2,4,6‑三苯基嘧啶类衍生物的合成方法与流程
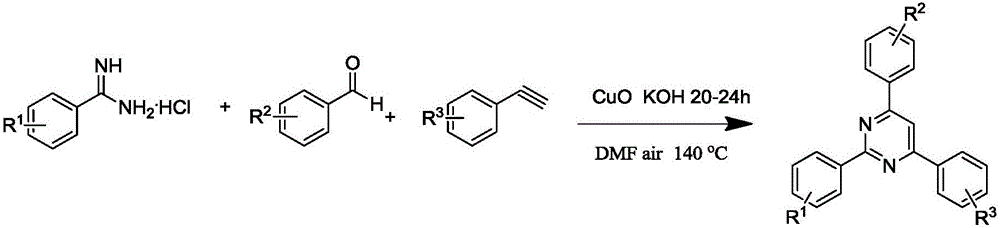
本发明属于有机合成方法领域,具体涉及一种2,4,6-三苯基嘧啶类衍生物的合成方法。
技术背景
嘧啶类化合物是指分子结构中含有两个氮原子的六元杂环化合物,与哒嗪、吡嗪互为同分异构体。嘧啶类化合物因具有不同的生物活性,如杀虫、杀菌、除草、抗病毒等。从而引起人们对这类化合物的广泛兴趣并进行了深入研究。例如维生素B、磺胺嘧啶、磺胺异二甲嘧啶、磺胺间二甲氧嘧啶;农药原料,如二嗪农、嘧啶醇、灭瘟素、除草定、环菌灵、甲菌定、乙菌定、磺嘧菌灵、比嗪农、抗蚜威、乙基虫螨磷、虫螨磷、多氧霉素、嘧啶威、嘧啶磷、氧嘧啶磷、嘧菌醇、异嘧菌醇、尿嘧啶等由于嘧啶化合物具有高效、对人类低毒等优点,因此其分子设计、合成与生物活性研究仍然是当今绿色农药创制的一个热点。
2,4,6-三苯基嘧啶是嘧啶环上的三个氢原子被其他原子或基团所取代,由于结构特点,在生物活性和药理性质方面有着广泛的应用,因此探索发展2,4,6-三苯基嘧啶的合成方法与技术任然是有机化学领域重要的研究课题之一。
目前文献中介绍的合成2,4,6-三苯基嘧啶的方法主要有以下几种:
(一)使用复杂的催化剂A催化苄脒盐酸盐和两组分的醇一锅法反应合成2,4,6-三苯基嘧啶衍生物。
该反应做到了三组分一锅法反应,且运用两种醇反应,符合绿色化学理念,但催化剂合成复杂,制约了其工业发展。
(二)使用五氯化铌(NbCl5)催化腈和炔反应合成2,4,6-三苯基嘧啶衍生物。
此法反应条件温和,但两组分腈制约嘧啶的取代基相同,且产率较低。
(三)2012年Jean-Marc Campagne课题组利用β-enaminones作为中间体和酰胺进行反应得到2,4,6-三取代嘧啶衍生物。
该合成方法需要对中间体进行保护基团保护,且反应分步进行,合成效率不高。
(四)以苄脒盐酸盐和丙炔醇类化合物在微波条件反应合成2,4,6-三取代嘧啶衍生物。
这种方法利用微波条件,条件简单但不利于工业扩大生产且反应过程使用MnO2重金属,污染环境。
(五)使用醋酸钯和碘化亚铜催化苄脒盐酸盐、碘苯、炔合成路线合成目标产物
但该合成方法使用了贵金属Pd做催化剂,三叔丁基磷做配体,且反应产率一般。
综上所述,现代合成2,4,6-三取代嘧啶化合物的方法很多,但有些合成方法4,6位取代基相同,不能做到不同取代基产物;有些使用贵金属钯作为催化剂,成本较高;有些合成步骤较为复杂或产率较低;因此提供一种合成高效,底物适用性广的新型合成方法十分必要。
技术实现要素:
针对现有技术上的不足,本发明旨在提供一种2,4,6-三苯基嘧啶类衍生物的合成方法,在空气条件下利用氧化铜催化苄脒盐酸盐、醛、炔三组分一锅法反应,合成方法成本低,原料易得,底物普适性高。
本发明提供的一种2,4,6-三苯基嘧啶类衍生物的合成方法,包括以下步骤:
A、在空气条件下,混合苄脒盐酸盐、醛和炔,然后加入催化剂、碱和溶剂,加热反应;
B、反应结束后,将产物分离、干燥、纯化后得到2,4,6-三苯基嘧啶类衍生物。
步骤A中所述苄脒盐酸盐、醛、炔、催化剂、碱的物质的量比为1:1.1-1.3:1.2-1.5:0.1:2-3。
步骤A中所述苄脒盐酸盐结构为:
式中R1为4-H、4-CH3、4-Cl、4-Br或4-CF3中的一种。
步骤A中所述醛结构为:
式中R2选自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3中的一种。
步骤A中所述炔结构为:
式中R3选自4-H、4-CH3、4-OCH3、4-Cl、4-Br或4-F中的一种。
步骤A中所述加热反应是指:在120℃-140℃条件下反应20-24小时;
步骤A中所述催化剂为氧化铜;所述碱为氢氧化钾、氢氧化钠、碳酸铯或碳酸钾中的一种;所述溶剂为N,N-二甲基甲酰胺、二甲基亚砜或N,N-二甲基乙酰胺。苄脒盐酸盐在溶剂中的浓度为0.1-0.5mol/L。
步骤B具体为:将所得产物用乙酸乙酯萃取,然后无水硫酸镁干燥,减压浓缩后,得粗产物,以体积比纯石油醚:乙酸乙酯=50:1为展开剂,通过柱层析分离得到2,4,6-三苯基嘧啶类衍生物。
步骤B所得2,4,6-三苯基嘧啶类衍生物结构式为式中,R1R1为4-H、4-CH3、4-Cl、4-Br或4-CF3;R2选自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3;R3选自4-H、4-CH3、4-OCH3、4-Cl、4-Br或4-F。
本发明中,首先醛与离去盐酸的脒进行了缩合反应生成亚胺,紧接着炔通过质子的转移与亚胺结合形成嘧啶中间体,最后在氧气的作用下脱H质子得到六元环2,4,6-嘧啶。本发明控制原料用量比,保证产率,以苄脒盐酸盐为反应基准,所以醛和炔是过量的,首先苄脒盐酸盐和醛进行缩合,所以1.2-1.3equiv的量可以使苄脒盐酸盐较为充分的转化,炔的量加到1.3-1.5equiv,是因为炔易挥发,也是确保最大程度的进行反应,保证最大化的产物收率。
与现有技术相比,本发明具有以下优点:(1)没有使用贵金属,有机氧化剂,而是利用商品化氧化铜作催化剂,节约成本;(2)利用苄脒盐酸盐、醛、炔为反应原料,原料易得;(3)该合成方法效率高,使用范围广,底物适用性广。
附图说明
图1为反应方程式;
式中,R1为4-H、4-CH3、4-Cl、3-Br或4-CF3;R2选自4-H、4-CH3、4-OCH3、4-Cl、3-Cl、2-Cl、4-Br、4-F或4-CF3;R3选自4-H、4-CH3、4-OCH3、4-Cl、4-Br、或4-F;
图2为实施例1核磁共振氢谱;
图3为实施例1核磁共振碳谱;
图4为实施例2核磁共振氢谱;
图5为实施例2核磁共振碳谱;
图6为实施例3核磁共振氢谱;
图7为实施例3核磁共振碳谱;
图8为实施例4核磁共振氢谱;
图9为实施例4核磁共振碳谱;
图10为实施例5核磁共振氢谱;
图11为实施例5核磁共振碳谱;
图12为实施例6核磁共振氢谱;
图13为实施例6核磁共振碳谱;
图14为实施例7核磁共振氢谱;
图15为实施例7核磁共振碳谱;
图16为实施例8核磁共振氢谱;
图17为实施例9核磁共振氢谱;
图18为实施例9核磁共振碳谱。
具体实施方式
实施例1
2,4,6-三苯基嘧啶的合成,包括以下步骤:
A、取0.3mmol苄脒盐酸盐和0.39mmol苯甲醛、0.45mmol苯乙炔混合,再往其加入0.03mmol CuO,0.9mmol KOH和2-3ml DMF,在140℃下搅拌反应24小时;
B、所得产物用乙酸乙酯萃取,然后无水硫酸镁干燥,减压浓缩后,得粗产物,用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即2,4,6-三苯基嘧啶,产率85%,熔点为182℃。
核磁表征数据:
1H NMR(300MHz,CDCl3):δ7.48-7.50(m,9H);7.97(s,1H),8.24–8.25(m,4H),8.67–8.69(m,2H);
13C NMR(75MHz,CDCl3):δ110.35,127.33,128.47,128.51,128.65,128.94,130.68,130.82,137.51,138.10,164.50,164.78。
实施例2
4-(4-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步骤:
A、0.3mmol对苄脒盐酸盐和0.36mmol对氯苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下搅拌反应24小时后,得产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体4-(4-氯苯基)-2,6-二苯基嘧啶。产率81%,165-167℃。
核磁表征数据:
1H NMR(300MHz,CDCl3):δ7.47-7.50(t,8H),7.79(s,1H),8.18-8.24(m,4H),8.66(t,J=3Hz,2H).
13C NMR(75MHz,CDCl3):δ109.91,127.29,128.49,128.95,129.13,135.89,136.97,137.31,137.95,163.42,164.51,164.89.
实施例3
2-(4-氯苯基)-4-苯基-6-(对甲苯基)嘧啶的合成,包括以下步骤:
A、取0.3mmol苄脒盐酸盐和0.39mmol对氯苯甲醛、0.40mmol对甲基苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下搅拌反应24小时;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即2-(4-氯苯基)-4-苯基-6-(对甲苯基)嘧啶,产率76%,熔点为164℃。
核磁表征数据:
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.30-7.33(d,J=9.0Hz,2H),7.48-7.49(d,J=3Hz,2H),7.90(s,1H),8.12-8.20(m,4H),8.64-8.65(d,J=3Hz,2H);
13C NMR(75MHz,CDCl3):δ109.63,127.20,128.48,128.57,129.13,129.69,130.71,134.51,136.04,136.89,138.04,141.34,163.35,164.48,164.88。
实施例4
4-(2-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步骤:
A、取1.5mmol苄脒盐酸盐和1.9mmol邻氯苯甲醛、2.25mmol苯乙炔,再往其加入0.15mmol CuO,4.5mmol KOH以及3-4ml DMF,在140℃下搅拌反应24小时得到产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即4-(2-氯苯基)-2,6-二苯基嘧啶。产率70%,熔点为124-125℃。
核磁表征数据:
1H NMR(300MHz,CDCl3):δ7.38-7.51(m,9H),7.79-7.81(t,1H),7.97(s,1H),8.23(d,J=3.9Hz,2H),8.62(d,J=3.6Hz,2H);
13C NMR(75MHz,CDCl3):δ115.19,127.26,127.40,128.08,128.50,128.97,130.52,130.73,130.93,131.72,132.45,137.26,137.56,137.93,163.90,164.69。
实施例5
4-(3-氯苯基)-2,6-二苯基嘧啶的合成,包括以下步骤:
A、取2mmol苄脒盐酸盐和2.6mmol间氯苯甲醛、3mmol苯乙炔,再往其加入0.2mmol CuO,6mmol KOH以及6-8ml DMF,在140℃下搅拌反应24小时后,得产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即4-(3-Cl苯基)-2,6-二苯基嘧啶,产率52%,熔点为147-148℃.
核磁表征数据:
1H NMR(300MHz,CDCl3):δ7.46-7.50(m,8H)7.94(s,1H),8.11(d,J=5.7Hz,1H,)8.24,(s,3H),8.67(d,J=4.8Hz,2H)
13C NMR(75MHz,CDCl3):δ110.218,125.34,127.31,127.42,128.51,128.66,128.96,130.16,130.71,130.83,130.98,135.08,137.23,137.87,139.33,163.24,164.57,164.98.
实施例6
4-(对甲苯基)-2,6-二苯基嘧啶的合成,包括以下步骤:
A、取3mmol苄脒盐酸盐和3.9mmol对甲基苯甲醛、4.5mmol苯乙炔,再往其加入0.3mmol CuO,9mmol KOH以及6-8ml DMF,在140℃下搅拌反应24小时,得到产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即4-(对甲苯基)-2,6-二苯基嘧啶,产率83.1%,熔点为152-154℃.
核磁表征数据:
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.32(d,J=7.5Hz),7.48-7.50(m,6H),7.50(s,1H),8.15(d,J=7.8Hz,2H),8.24(d,J=5Hz),8.67(d,J=6Hz).
13C NMR(75MHz,CDCl3):δ21.51,109.99,127.23,127.32,128.44,128.50,128.91,129.67,130.60,130.72,134.70,137.64,138.23,141.18,164.42,164.62,164.69.
实施例7
2,4-二苯基-6-(2-噻吩基)嘧啶的合成,包括以下步骤:
A、取0.15mmol苄脒盐酸盐和0.18mmol 2-噻吩甲醛、0.225mmol苯乙炔,再往其加入0.015mmol CuO,0.45mmol KOH以及1-2ml DMF,在120℃下搅拌反应24小时得到产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体,2,4-二苯基-6-(2-噻吩基),嘧啶产率86.3%,熔点为169-172℃
1H NMR(300MHz,CDCl3):δ7.17(d,J=3.6Hz,2H),7.48-7.50(m,6H),7.81(s,1H),7.89(s,1H),8.21(d,J=3.9Hz,2H),8.63(d,J=4.5Hz 2H).
13C NMR(75MHz,CDCl3):δ108.44,127.09,127.26,128.33,128.47,128.63,128.92,129.82,130.76,130.85,137.34,137.77,143.39,159.70,164.45,164.56.
实施例8
4,6-二苯基-2-(对甲苯基)嘧啶的合成,包括以下步骤:
A、0.3mmol对甲基苄脒盐酸盐和0.39mmol苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下搅拌反应24小时,得到产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体即4,6-二苯基-2-(对甲苯基)嘧啶。产率为88%,熔点为182-185℃
1H NMR(300MHz,CDCl3):δ2.41(s,3H),7.30(d,J=7.8Hz),7.50-7.52(d,6H),7.94(s,1H),8.24(t,J=5.4Hz,4H),8.58(d,J=7.8Hz,2H);
13C NMR(75MHz,CDCl3):δ21.60,110.06,127.31,128.48,128.91,129.94,130.72,135.51,137.67,140.85,164.67.
实施例9
4,6-二苯基-2-(对三氟甲基)嘧啶的合成,包括以下步骤:
A、0.3mmol对三氟甲基苄脒盐酸盐和0.39mmol苯甲醛、0.45mmol苯乙炔,再往其加入0.03mmol CuO,0.9mmol KOH以及2-3ml DMF,在140℃下搅拌反应24小时,得到产物;
B、将产物用乙酸乙酯进行萃取,干燥得到粗产物,将粗产物用硅胶柱层析(溶剂体积比为石油醚:乙酸乙酯=50:1)纯化得白色固体4,6-二苯基-2-(对三氟甲基)嘧啶。产率为84.5%,熔点197-199℃
1H NMR(500MHz,CDCl3):δ7.58-7.61(m,6H),7.79(d,2H,8.5Hz),8.08(s,1H),8.29(t,4H),8.84(d,2H,8Hz).
13C NMR(75MHz,CDCl3):δ110.94,125.34,125.39,127.29,128.74,129.01,131.03,132.37,137.16,141.41,163.17,164.95.
以上详细描述了本发明的优选实施方式。但是,本发明并不限于上述实施方式中的具体细节。只要在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型的均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!