医用水凝胶前体及其制备方法和医用水凝胶以及应用与流程
本申请涉及一种医用水凝胶前体及其制备方法和医用水凝胶以及应用,属于生物医学领域。
背景技术:
水凝胶是一种具有三维网络结构的亲水性聚合物,具有高含水量和柔软特性,在生物医学领域具有广泛的应用。现有的医用水凝胶主要包括天然聚合物类产品、丙烯酰胺类产品和聚乙二醇(PEG)类产品。其中,PEG类医用水凝胶以其良好的生物相容性、可降解性等特点在药物缓释、组织工程、组织粘合封闭等方面有很好的应用前景。
现有技术中,基于迈克尔加成机理的医用水凝胶均以聚醚结构为材料主体结构单元,例如DURESEAL、HYPERBRANCH等公司的产品,均是以PEG材料为主体成分,PEG端基修饰后作为医用水凝胶的制备原料。但是现有的PEG类医用水凝胶其制备原料的合成工艺复杂,市场销售价格较高,对患者造成了比较大的经济压力。
技术实现要素:
发明要解决的问题
本申请的目的在于提供一种医用水凝胶前体及其制备方法和医用水凝胶以及应用。该医用水凝胶前体能够用于制备医用水凝胶和可负载活体细胞的生物支架。
进一步地,医用水凝胶前体的制备方法简单,成本较低。
用于解决问题的方案
本申请提供一种医用水凝胶前体,所述医用水凝胶前体包括如下式(1)所示的化合物:
其中,R表示多元醇中除去n个羟基而得到的基团;A、B表示碳原子数为2-6的亚烷基;R’表示活性基团,所述活性基团含有N-琥珀酰亚胺基、氨基或巯基中的一种;n≥3,k≥1,m≥1,且n、k、m为整数。
根据本申请的医用水凝胶前体,所述亚烷基选自如下式(2)或式(3)所示的基团:
其中,R1表示氢原子或者碳原子数为1-4的烷基。
根据本申请的医用水凝胶前体,所述A选自下式(4)或式(5)所示的基团:
根据本申请的医用水凝胶前体,所述B为如下式(6)所示的基团:
-CH2-CH2- (6)。
根据本申请的医用水凝胶前体,所述多元醇包括丙三醇、三羟甲基乙烷、季戊四醇、木糖醇、山梨糖醇中的一种。
本申请还提供一种根据本申请的医用水凝胶前体的制备方法,包括以下步骤:
聚合步骤:在无水无氧,且氮气存在的环境下,将多元醇与碳原子数为2-6的环氧化物进行阴离子开环聚合反应,得到星形共聚物;
修饰步骤:利用活性基团对所述星形共聚物进行修饰,得到医用水凝胶前体;所述活性基团含有N-琥珀酰亚胺基,氨基或巯基中的一种。
根据本申请的医用水凝胶前体的制备方法,所述聚合步骤包括:
第一聚合步骤:在无水无氧,且氮气存在的环境下,将多元醇与碳原子数为2-6的第一环氧化物进行阴离子开环聚合反应,得到星形共聚物前体;
第二聚合步骤:在无水无氧,且氮气存在的环境下,将所述星形共聚物前体与碳原子数为2-6的第二环氧化物进行阴离子开环聚合反应,得到所述星形共聚物。
根据本申请的医用水凝胶前体的制备方法,所述第一环氧化物为环氧丙烷;所述第二环氧化物为环氧乙烷。
根据本申请的医用水凝胶前体的制备方法,在一种具体的实施方式中,所述修饰步骤包括:
将所述星形共聚物与亚硫酰二溴置于甲苯和三乙胺溶液中反应,得到修饰有溴原子的星形共聚物;
将所述修饰有溴原子的星形共聚物与乙醇和氨气进行反应,得到修饰有氨基的星形共聚物,或者
在催化剂的存在下,将所述修饰有溴原子的星形共聚物与叠氮钠反应,得到修饰有氨基的星形共聚物。
根据本申请的医用水凝胶前体的制备方法,在本实施方式中,所述修饰步骤还包括:
在碳二亚胺类缩合剂和酰化催化剂存在的条件下,将修饰有氨基的星形共聚物与巯基化试剂反应,得到修饰有巯基的星形共聚物;
优选地,所述巯基化试剂包括半胱氨酸、乙酰基半胱氨酸、巯基乙酸、巯基丙酸中的一种或多种;
更优选地,所述碳二亚胺类缩合剂包括1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐及其衍生物、N,N'-二异丙基碳二亚胺及其衍生物、二环己基碳二亚胺及其衍生物中的一种或多种;
进一步优选地,所述酰化催化剂包括4-二甲氨基吡啶及其衍生物、4-吡咯烷基吡啶及其衍生物、羟基苯并三唑及其衍生物、N-羟基琥珀酰亚胺及其衍生物、N-羟基硫代琥珀酰亚胺及其衍生物中的一种或多种。
根据本申请的医用水凝胶前体的制备方法,在另一种具体的实施方式中,所述修饰步骤包括:
羧基引入步骤:在缚酸剂的存在下,将所述星形共聚物与酸酐类化合物反应,得到修饰有羧基的星形共聚物;
羧基活化步骤:在碳二亚胺类缩合剂和酰化试剂存在的条件下,将所述修饰有羧基的星形共聚物中的羧基活化为羧基活性酰胺,得到修饰有N-琥珀酰亚胺基的星形共聚物;
优选地,所述酸酐类化合物包括丁二酸酐、戊二酸酐、己二酸酐中的一种或两种以上的组合;
更优选地,所述缚酸剂包括三乙胺、吡啶中一种或两种;
再优选地,所述碳二亚胺类缩合剂包括1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐及其衍生物、N,N'-二异丙基碳二亚胺及其衍生物、二环己基碳二亚胺及其衍生物中的一种或多种;
进一步优选地,所述酰化试剂包括4-二甲氨基吡啶及其衍生物、4-吡咯烷基吡啶及其衍生物、羟基苯并三唑及其衍生物、N-羟基琥珀酰亚胺及其衍生物、N-羟基硫代琥珀酰亚胺及其衍生物中的一种或多种。
本申请还提供一种医用水凝胶前体组合物,所述医用水凝胶前体组合物包含(A)含有氨基和/或巯基的医用水凝胶前体的第一组分,和(B)含有N-琥珀酰亚胺基的医用水凝胶前体的第二组分;
其中所述含有氨基和/或巯基的医用水凝胶前体是本申请的医用水凝胶前体中活性基团含有氨基和/或巯基的医用水凝胶前体;
所述含有N-琥珀酰亚胺基的医用水凝胶前体是本申请的医用水凝胶前体中活性基团含有N-琥珀酰亚胺基的医用水凝胶前体。
本申请还提供一种医用水凝胶,所述医用水凝胶通过使用本申请的含有氨基和/或巯基的医用水凝胶前体与所述的含有N-琥珀酰亚胺基的医用水凝胶前体反应后形成。
根据本申请的医用水凝胶,由所述含有氨基和/或巯基的医用水凝胶前体溶于第一缓冲溶液中,所述含有N-琥珀酰亚胺基的医用水凝胶前体溶于第二缓冲溶液中,然后使两种溶液混合、反应后形成。
本申请还提供一种可负载活体细胞的生物支架,包括本申请的含有氨基和/或巯基的医用水凝胶前体与所述的含有N-琥珀酰亚胺基的医用水凝胶前体反应后形成。
本申请还提供一种根据本申请的医用水凝胶的制备方法,包括以下步骤:
将所述含有氨基和/或巯基的医用水凝胶前体溶于第一缓冲溶液中,得到第一混合液;
将所述含有N-琥珀酰亚胺基的医用水凝胶前体溶于第二缓冲溶液中,得到第二混合液;
将所述第一混合液与第二混合液混合,得到所述医用水凝胶。
根据本申请的医用水凝胶的制备方法,所述第一缓冲溶液和/或第二缓冲溶液包括磷酸盐溶液、碳酸盐溶液、硼酸盐溶液、磷酸溶液、乙酸溶液、盐酸溶液中一种或多种。
本申请还提供一种医用水凝胶试剂盒,包括本申请的含有氨基和/或巯基的医用水凝胶前体、含有N-琥珀酰亚胺基的医用水凝胶前体以及用于溶解所述医用水凝胶前体的缓冲溶液。
本申请还提供一种根据本申请的医用水凝胶在制备伤口粘合制品、内脏或软组织伤口闭合制品、脑脊液及组织液封堵制品、大面积创伤止血辅助物、软组织修复补片或软组织粘合制品中的应用。
本申请还提供一种根据本申请的可负载活体细胞的生物支架在细胞培养中的应用。
发明的效果
通过采用本申请的医用水凝胶前体所制备得到的医用水凝胶具有良好的粘附强度,可以使组织创面快速闭合,能够有效起到组织封闭的作用,防止组织液或血液或脑脊液泄漏,且所述医用水凝胶前体可以完全生物降解,从而避免二次手术,减少病人痛苦,还能够降低治疗费用。
进一步地,通过采用本申请的医用水凝胶前体所制备得到的可负载活体细胞的生物支架能够为细胞的分裂增殖提供一个良好的生长环境和三维空间,浸润在培养基中可使细胞在生物支架存在的环境中增殖。
附图说明
图1为实验组T2接受硬脑膜修补术后,将医用水凝胶IV喷涂于人工硬脑膜周围后的照片。
图2为医用水凝胶II应用于自体硬脑膜修复部位的组织切片图。
图3为细胞毒实验中,实验组、对照组分别减去空白组后(即以空白为参照)的吸光度值(OD值)。
图4为采用实施例3的医用水凝胶前体经生物3D打印后的生物支架的光学照片。
图5为负载骨髓间质干细胞的生物支架的显微镜照片。
图6为生物支架的场发射电子扫描显微镜照片。
具体实施方式
本申请一种医用水凝胶前体,所述医用水凝胶前体包括如下式(1)所示的化合物:
其中,R表示多元醇中除去n个羟基而得到的基团;A、B表示碳原子数为2-6的亚烷基;R’表示活性基团,所述活性基团含有N-琥珀酰亚胺基、氨基或巯基中的一种;n≥3,k≥1,m≥1,且n、k、m为整数。
优选地,n为3-6之间的整数,更优选地,n为3或4;k为1-10之间的整数,m为1-10之间的整数;更优选地,k与m之和在5-10的范围内。此时所述医用水凝胶前体具有较合适的分子量,可以使制得的医用水凝胶具有适宜的降解效果。
当R’为含有N-琥珀酰亚胺基的基团时,所述R’可以为-R”NHS,其中,所述R”可以不存在,即R’为-NHS;
或者所述R”可以为-OC(CH2)x-、-OC(CH2)xCO-、-CH2CO-、-(CH2)xNHOC(CH2)xCO-或-(CH2)xNHOC(CH2)x+1CO-;其中,x为0-10之间的整数,优选0-5之间的整数,更优选为0-3之间的整数。
当R’为含有氨基或巯基的基团时,所述R’可以为-R0SH或-R0NH2,其中,所述R0可以为-(CH2)y-,y可以为0-10之间的整数,优选0-5之间的整数,更优选0-2之间的整数;当y为0时,所述R0不存在,即R’为-SH或-NH2。
本申请的医用水凝胶前体是修饰有活性基团的星形共聚物,(即:多臂共聚物),具有多臂聚醚结构。在使用时,将修饰有N-琥珀酰亚胺基的医用水凝胶前体与修饰有氨基和/或巯基的医用水凝胶前体物理混合,从而原位形成医用水凝胶。
优选地,所述亚烷基选自如下式(2)或式(3)所示的基团:
其中,R1表示氢原子或者碳原子数为1-4的烷基。
根据本申请的医用水凝胶前体,其中,所述多元醇包括丙三醇、三羟甲基乙烷、季戊四醇、木糖醇、山梨糖醇中的一种。
根据本申请的医用水凝胶前体,其中,所述A选自下式(4)或式(5)所示的基团:
所述B为如下式(6)所示的基团:
-CH2-CH2- (6)。
本申请还提供一种根据本申请的医用水凝胶前体的制备方法,包括以下步骤:
聚合步骤:在无水无氧,且氮气存在的环境下,将多元醇与碳原子数为2-6的环氧化物进行阴离子开环聚合反应,得到星形共聚物;
修饰步骤:利用活性基团对所述星形共聚物进行修饰,得到医用水凝胶前体,即
其中,R表示多元醇中除去n个羟基而得到的基团;A、B表示碳原子数为2-6的亚烷基;R’表示活性基团,所述活性基团含有N-琥珀酰亚胺基、氨基或巯基中的一种;n≥3,k≥1,m≥1,且n、k、m为整数。
优选地,n为3-6之间的整数,更优选地,n为3或4;k为1-10之间的整数,m为1-10之间的整数;优选地,k与m之和在5-10的范围内。此时所述医用水凝胶前体具有较合适的分子量,可以使制得的医用水凝胶具有适宜的降解效果。
当R’为含有N-琥珀酰亚胺基的基团时,所述R’可以为-R”NHS,其中,所述R”可以不存在,即R’为-NHS;
或者所述R”可以为-OC(CH2)x-、-OC(CH2)xCO-、-CH2CO-、-(CH2)xNHOC(CH2)xCO-或-(CH2)xNHOC(CH2)x+1CO-;其中,x为0-10之间的整数,优选0-5之间的整数,更优选为0-3之间的整数。
当R’为含有氨基或巯基的基团时,所述R’可以为-R0SH或-R0NH2,其中,
所述R0可以为-(CH2)y-,y可以为0-10之间的整数,优选0-5之间的整数,更优选0-2之间的整数;当y为0时,所述R0不存在,即R’为-SH或-NH2。
优选地,当本申请中的A、B不相同时,所制备得到的星形共聚物为星形嵌段共聚物。
优选地,所述多元醇为丙三醇三羟甲基乙烷季戊四醇木糖醇山梨糖醇等。
根据本申请的医用水凝胶前体的制备方法,所述聚合步骤包括:
第一聚合步骤:在无水无氧,且氮气存在的环境下,将多元醇与碳原子数为2-6的第一环氧化物进行阴离子开环聚合反应,得到星形共聚物前体;
第二聚合步骤:在无水无氧,且氮气存在的环境下,将所述星形共聚物前体与碳原子数为2-6的第二环氧化物进行阴离子开环聚合反应,得到所述星形共聚物。
优选地,当所述第一环氧化物与所述第二环氧化物不相同时,所制备得到的星形共聚物前体为星形嵌段共聚物前体,所制备得到的星形共聚物为星形嵌段共聚物。
具体地,在第一聚合步骤中的反应温度在多元醇的凝固点以上,碳原子数为2-6的第一环氧化物的沸点以下。在第二聚合步骤中的反应温度在碳原子数为2-6的第二环氧化物的沸点以下。可以通过加入氢氧化物、醇盐、金属氧化物、金属有机化合物或其他阴离子活性物作为引发剂从而引发阴离子开环聚合反应。
根据本申请的医用水凝胶前体的制备方法,所述第一环氧化物为环氧丙烷;所述第二环氧化物为环氧乙烷。
举例而言,丙三醇的凝固点为17.8℃,环氧丙烷的沸点为34℃,当采用丙三醇与环氧丙烷进行第一聚合步骤时,则反应温度需在17.8℃~34℃之间。而环氧乙烷的沸点为10.4℃,当采用星形共聚物前体与环氧乙烷进行第二聚合步骤时,则反应温度需低于10.4℃。
另外,当采用其它多元醇和其它第一环氧化物、第二环氧化物进行反应时,反应温度的控制与采用丙三醇和环氧丙烷、环氧乙烷进行反应的方式相同。
根据本申请的医用水凝胶前体的制备方法,在一种具体的实施方式中,所述修饰步骤包括:
将星形共聚物与亚硫酰二溴置于甲苯和三乙胺溶液中反应,得到修饰有溴原子的星形共聚物;
将所述修饰有溴原子的星形共聚物与乙醇和氨气进行反应,得到修饰有氨基的星形共聚物,或者
将所述修饰有溴原子的星形共聚物与叠氮钠在催化剂的存在下进行反应,得到修饰有氨基的星形共聚物;其中,所述催化剂为Pd/C,且所述修饰有溴原子的星形共聚物与叠氮钠在二甲基甲酰胺(DMF)溶液中混合并进行反应。
根据本申请的医用水凝胶前体的制备方法,在本实施方式中,所述修饰步骤还包括:
在碳二亚胺类缩合剂和酰化催化剂存在的条件下,将修饰有氨基的星形共聚物与巯基化试剂反应,得到修饰有巯基的星形共聚物;
优选地,所述巯基化试剂包括但不限于:半胱氨酸、乙酰基半胱氨酸、巯基乙酸、巯基丙酸中的一种或多种;
更优选地,所述的碳二亚胺类缩合剂包括但不限于:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)或其衍生物、N,N'-二异丙基碳二亚胺(DIC)或其衍生物、二环己基碳二亚胺(DCC)或其衍生物中的一种或多种。
进一步优选地,所述酰化催化剂包括但不限于:4-二甲氨基吡啶及其衍生物、4-吡咯烷基吡啶及其衍生物、羟基苯并三唑及其衍生物、N-羟基琥珀酰亚胺及其衍生物、N-羟基硫代琥珀酰亚胺及其衍生物中的一种或多种。
根据本申请的医用水凝胶前体的制备方法,在另一种具体的实施方式中,所述修饰步骤包括:
羧基引入步骤:在缚酸剂的存在下,将所述星形共聚物与酸酐类化合物反应,得到修饰有羧基的星形共聚物;
羧基活化步骤:在碳二亚胺类缩合剂和酰化试剂存在的条件下,将所述修饰有羧基的星形共聚物中的羧基活化为羧基活性酰胺,得到修饰有N-琥珀酰亚胺基的星形共聚物。
优选地,所述酸酐类化合物包括但不限于:丁二酸酐、戊二酸酐、己二酸酐中的一种或两种以上的组合,优选为丁二酸酐;其中,戊二酸酐、己二酸酐具有一定的毒性,在终产物中可以除去。
更优选地,所述缚酸剂包括但不限于:三乙胺、吡啶中的一种或两种。
再优选地,所述的碳二亚胺类缩合剂包括但不限于:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)或其衍生物、N,N'-二异丙基碳二亚胺(DIC)或其衍生物、二环己基碳二亚胺(DCC)或其衍生物中的一种或多种。另外,制备含氨基或巯基的医用水凝胶前体与制备含N-琥珀酰亚胺基的医用水凝胶前体所使用的碳二亚胺类缩合剂可以相同也可以不同。
进一步优选地,所述酰化试剂包括但不限于:4-二甲氨基吡啶及其衍生物、4-吡咯烷基吡啶及其衍生物、羟基苯并三唑及其衍生物、N-羟基琥珀酰亚胺及其衍生物、N-羟基硫代琥珀酰亚胺及其衍生物中的一种或多种。
本申请还提供一种医用水凝胶前体组合物,所述医用水凝胶前体组合物包含(A)含有氨基和/或巯基的医用水凝胶前体的第一组分,和(B)含有N-琥珀酰亚胺基的医用水凝胶前体的第二组分;
其中,所述含有氨基和/或巯基的医用水凝胶前体是本申请的医用水凝胶前体中活性基团含有氨基和/或巯基的医用水凝胶前体。
所述含有N-琥珀酰亚胺基的医用水凝胶前体是本申请的医用水凝胶前体中活性基团含有N-琥珀酰亚胺基的医用水凝胶前体。
本申请还提供一种医用水凝胶,包括本申请的含有氨基和/或巯基的医用水凝胶前体与含有N-琥珀酰亚胺基的医用水凝胶前体反应后形成。一般而言,所述含有氨基和/或巯基的医用水凝胶前体为亲核试剂,所述含有N-琥珀酰亚胺基的医用水凝胶前体为亲电试剂。
根据本申请的医用水凝胶,由所述含有氨基和/或巯基的医用水凝胶前体(亲核试剂)溶于第一缓冲溶液中,所述含有N-琥珀酰亚胺基的医用水凝胶前体(亲电试剂)溶于第二缓冲溶液中,然后混合、反应后形成。
通过采用本申请的医用水凝胶前体制备得到的医用水凝胶,具有良好的粘附强度,可以使组织创面快速闭合,有效起到组织封闭的作用,防止组织液或血液或脑脊液泄漏。还可以完全生物降解,避免二次手术,减少病人痛苦。
本申请还提供一种根据本申请的医用水凝胶的制备方法,包括以下步骤:
将所述含有氨基和/或巯基的医用水凝胶前体(亲核试剂)溶于第一缓冲溶液中,得到第一混合液;
将所述含有N-琥珀酰亚胺基的医用水凝胶前体(亲电试剂)溶于第二缓冲溶液中,得到第二混合液;
将所述第一混合液与第二混合液混合,得到所述医用水凝胶。
本申请的医用水凝胶的制备方法,所述第一缓冲溶液和/或第二缓冲溶液包括但不限于:磷酸盐溶液、碳酸盐溶液、硼酸盐溶液、磷酸溶液、乙酸溶液、盐酸溶液中一种或多种。所述缓冲溶液的具体成分可根据亲核试剂或亲电试剂在缓冲溶液中的稳定性和成型的医用水凝胶的理化性能情况进行选择,缓冲溶液不应含有有害或有毒的溶剂,通常选用水做溶剂,缓冲液的渗透压应与生物体血液渗透压相同或接近。
优选地,所述第一缓冲溶液包括但不限于:pH值为8.0-10.0的四硼酸钠缓冲溶液或pH为9.6~9.9的磷酸二氢钠溶液与碳酸钠溶液的组合的缓冲溶液;所述第二缓冲溶液包括但不限于:pH值为3.5-5.0的盐酸溶液、pH为5.0~6.0的磷酸钠缓冲溶液或pH为3.5-4.5的磷酸二氢钠溶液和碳酸溶液的组合的缓冲溶液。
本申请的医用水凝胶的制备方法,所述第一缓冲溶液液和/或第二缓冲溶液中包含有显色剂;优选地,所述显色剂包括但不限于:FD&C蓝色#1、FD&C蓝色#2、FD&C蓝色#3、D&C绿色#6、亚甲基蓝中的一种或多种。
本申请还提供一种可负载活体细胞的生物支架,包括本申请的含有氨基和/或巯基的医用水凝胶前体与含有N-琥珀酰亚胺基的医用水凝胶前体反应后形成。
通过采用本申请的医用水凝胶前体制备得到的可负载细胞的生物支架,可以为细胞的分裂增殖提供一个良好的生长环境和三维空间,浸润在培养基中可使细胞在生物支架存在的环境中分裂增殖,通过负载不同细胞或不同细胞诱导因子,培养所需要的活体细胞。
本申请还提供一种根据本申请的可负载活体细胞的生物支架的制备方法,包括以下步骤:
将所述含有氨基和/或巯基的医用水凝胶前体(亲核试剂)溶于第三缓冲溶液中,得到第一混合液;
将所述含有N-琥珀酰亚胺基的医用水凝胶前体(亲电试剂)溶于第二缓冲溶液中,得到第二混合液;
将所述第一混合液与第二混合液混合,得到所述可负载活体细胞的生物支架;
其中,所述第三缓冲溶液包括但不限于pH为7.5~8.5的磷酸二氢钠溶液与碳酸钠溶液的组合的缓冲溶液。
通过采用医用水凝胶前体所制备得到的可负载活体细胞的生物支架能够为细胞的分裂增殖提供一个良好的生长环境和三维空间,浸润在培养基中可使细胞在生物支架存在的环境中增殖,通过负载不同细胞或不同细胞诱导因子,培养所需要的活体细胞。
本申请还提供一种医用水凝胶试剂盒,包括根据本申请的含有氨基和/或巯基的医用水凝胶前体(亲核试剂)、含有N-琥珀酰亚胺基的医用水凝胶前体(亲电试剂)以及用于溶液所述医用水凝胶前体的缓冲溶液。
本申请还提供一种根据本申请的医用水凝胶在伤口粘合制品、内脏或软组织伤口闭合制品、脑脊液及组织液封堵制品、大面积创伤止血辅助物、软组织修复补片与软组织粘合制品中的应用。
本申请还提供一种根据本申请的可负载活体细胞的生物支架在细胞培养中的应用。
另外,本申请中的成胶时间的测定方法为:将所述第一混合溶液与第二混合溶液混合后开始计时,当混合后的二元混合物不再流动时即为成胶,此段时间记为成胶时间。
本申请中的溶胀率的测试方法为:将制得的医用水凝胶置于精确度为万分之一位的天平上,测得其重量为x g。然后将所述医用水凝胶置于pH=7.4的磷酸盐(PBS)缓冲溶液中12小时,使得医用水凝胶达到溶胀平衡后取出。用滤纸擦干表面水分,再次将其置于精确度为万分之一位的天平上,测得其重量为yg。重复测定十次,计算其平均值x和y。按照以下公式计算溶胀率:
溶胀率=(y-x)/x×100%。
本申请中破裂强度的测定方法为:取新鲜猪皮或猪肠衣,其面积为20×20cm,将其固定于只有一个开口的密闭箱的开口处,然后将所述密闭箱的开口密封,密闭箱的另一侧连接有压力打气泵,并配有压力传感器。在所述猪皮或猪肠衣的中心位置切开一个直径为0.5cm±0.02cm的小口,然后将人工硬脑膜或其他可封堵的材料置于小口表面,通过双联注射器在封堵材料周围均匀的喷上所述第一混合溶液和所述第二混合溶液,喷涂后计时1至5分钟,然后通过压力泵向密闭箱内打气,记录人工硬脑膜或其他封堵材料被顶开时压力传感器的读数,即破裂强度。
本申请中的降解时间的测定方法为:称取一定重量的医用水凝胶(例如0.5g),置于不可降解的聚丙烯网袋中,将其充分干燥后,放入pH=7.4的磷酸盐(PBS)缓冲溶液中,然后置于恒温恒湿的37℃烘箱内。每隔24小时从缓冲溶液中取出一次样品,吸干其表面水分并干燥样品后测试其重量。重量损失超过其原始重量5%时记为开始降解时间;重量损失达到100%时为完全降解时间。
实施例
下面将结合实施例对本申请的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本申请,而不应视为限定本申请的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
巯基丙酸:Aladdin公司;产品编号:M103035。
可溶性碳二亚胺:Sigma-Aldrich公司;产品编号:MFCD00044916。
N-羟基琥珀酰亚胺(N-羟基丁二酰亚胺):Sigma-Aldrich公司;产品编号:56480。
3-arm-PEG-NH2:厦门赛诺邦格生物科技有限公司。
4-arm-PEG-NHS:厦门赛诺邦格生物科技有限公司。
4-arm-PEG-NH2:厦门赛诺邦格生物科技有限公司。
实施例1
<医用水凝胶前体的制备>
以丙三醇(10mmol,0.9209g,0.7303mL)作为起始剂,加入至100mL的反应瓶中,然后加入20mL的二氧六环,置于60℃水浴中,抽真空,并通入高纯氮气,反复操作,最后连续抽真空状态下保持2小时,以除去水和氧气。
将温度降至20℃,并持续通入高纯氮气,向体系内加入环氧丙烷(90mmol,5.2272g,6.2979mL)、甲醇钠(0.1mmol,0.0054g,0.0042mL)引发环氧丙烷的开环聚合反应,反应2小时。
将温度降至5℃,持续通入高纯氮气,然后加入适量环氧乙烷单体(180mmol,7.9294g,9.1027mL),持续反应2小时,通过减压蒸馏方式除去二氧六环,经纯化后,得到羟基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物,产物质量为10.7834g,产率为76.6%。
取羟基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(5.3917g,3.83mmol)与亚硫酰二溴(SOBr2,11.49mmol,2.3884g)在50mL二氯甲烷和三乙胺(17.25mmol,1.7455g,2.3911mL)中反应得到溴封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.9848g。
将溴封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物与叠氮钠(17.25mmol,1.1214g)在50mL的DMF溶剂中混合,以0.2g的Pd/C为催化剂,在20mL的乙醇溶剂中催化加氢,得到氨基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲核试剂),纯化后称重,质量为4.6658g。
取羟基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(5.3917g,3.83mmol)与丁二酸酐(17.235mmol,1.7247g)在50mL二氯甲烷和三乙胺(25.8525mmol,2.6160g,3.5836mL)中进行反应,制备得到羧基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.4784g。
将羧基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.4784g)与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(11.49mmol,2.2026g)和酰化试剂N-羟基琥珀酰亚胺(13.788mmol,1.3662g)于0℃的水溶液中连续搅拌反应40min,得到N-羟基琥珀酰亚胺基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲电试剂),纯化后称重,质量为4.0845g。
<医用水凝胶的制备>
将氨基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,1.4230g)溶于2.5mL的磷酸二氢钠与碳酸钠混合的缓冲溶液(pH=9.97±0.02)中,得到第一组分;将N-羟基琥珀酰亚胺基封端的丙三醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,2.0070g)溶于2.5mL磷酸钠缓冲溶液(pH=5.64±0.02)中,得到第二组分;将第一组分和第二组分通过挤出装置等体积混合挤出,得到医用水凝胶I。
实施例2
<医用水凝胶前体的制备>
以季戊四醇(10mmol,1.3615g)为起始剂,加入至100mL的反应瓶中,加入20mL的吡啶,置于60℃水浴中,抽真空,并通入高纯氮气,反复操作,最后连续抽真空状态下保持2小时,以除去水和氧气。
将温度降至20℃,并持续通入高纯氮气,加入环氧丙烷(80mmol,4.6464g,5.5981mL)、甲醇钠(0.1mmol,0.0054g,0.0042mL)引发环氧丙烷开环反应,并持续反应2小时。
将温度降至5℃,持续通入高纯氮气,然后加入环氧乙烷单体(160mmol,7.0483g,8.0913mL),持续反应2小时,通过减压蒸馏方式除去吡啶,经纯化后,得到羟基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物,产物质量为9.4527g,产率为72.4%。
取羟基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.7263g,3.62mmol)与亚硫酰二溴(14.48mmol,3.0110g)在二氯甲烷(50mL)和三乙胺(21.72mmol,2.1978g,3.0108mL)中反应得到溴封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.6258g。
将溴封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.6258g)与叠氮钠(21.72mmol,1.4120g)在50mL的DMF溶剂中混合,以0.2g的Pd/C为催化剂,在20mL的乙醇溶剂中催化加氢,得到氨基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲核试剂),纯化后称重,质量为4.0749g。
取羟基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.7263g,3.62mmol)与丁二酸酐(21.72mmol,2.1735g)在50mL二氯甲烷和三乙胺(32.58mmol,3.2968g,4.5161mL)中反应,制备得到羧基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.5474g。
将羧基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.5474g)与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(14.48mmol,2.7758g)和N-羟基琥珀酰亚胺(17.376mmol,1.7218g)于10℃的水溶液中连续搅拌反应20min,得到N-羟基琥珀酰亚胺基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲电试剂),纯化后称重,质量为4.1255g。
<医用水凝胶的制备>
将氨基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,1.3696g)溶于2.5mL磷酸二氢钠与碳酸钠混合的缓冲溶液(pH=9.97±0.02)中,得到第一组分;将N-羟基琥珀酰亚胺基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,2.1056g)溶于2.5mL磷酸二氢钠与碳酸混合的缓冲溶液(pH=3.64±0.02)中,得到第二组分;将第一组分和第二组分通过挤出装置等体积混合挤出,得到医用水凝胶II。
实施例3
<医用水凝胶前体的制备>
以山梨糖醇(6mmol,1.0930g)为起始剂,加入至100mL的反应瓶中,加入20mL二氧六环,置于60℃水浴中,抽真空,通入高纯氮气,反复操作,最后连续抽真空状态下保持2小时,以除去水和氧气。
将温度降至20℃,并持续通入高纯氮气,加入环氧丙烷(72mmol,4.1818g,5.0383mL)和甲醇钠(0.05mmol,0.0027g,0.0021mL)引发环氧丙烷开环反应,并持续反应2小时。
将温度降至5℃,持续通入高纯氮气,然后加入环氧乙烷单体(216mmol,9.5152g,10.9232mL),持续反应2小时,通过减压蒸馏方式除去二氧六环,经纯化后;得到羟基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,产物质量为10.2123g,产率为69.05%。
取羟基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(5.1062g,2.07mmol)与亚硫酰二溴(12.42mmol,2.5817g)在二氯甲烷(50mL)和三乙胺(18.63mmol,1.8852g,2.5824mL)中反应得到溴封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.8345g。
将溴封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.8345g)与叠氮钠(18.63mmol,1.2111g)在50mL的DMF溶剂中混合,以0.2g的Pd/C为催化剂,在20mL的乙醇溶剂中催化加氢,得到氨基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.1125g。
取羟基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(5.1062g,2.07mmol)与丁二酸酐(18.63mmol,1.8643g)在50mL的二氯甲烷和三乙胺(27.945mmol,2.8278g,3.8736mL)中反应,制备得到羧基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.5566g。
将羧基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.5566g)与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(12.42mmol,2.3809g)、N-羟基琥珀酰亚胺(14.904mmol,1.4768g)于5℃的水溶液中连续搅拌反应15min,得到N-羟基琥珀酰亚胺基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.0756g。
<医用水凝胶的制备>
将氨基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,2.5610g)溶于2.5mL磷酸二氢钠与碳酸钠混合的缓冲溶液(pH=9.97±0.02)中,得到第一组分;将N-羟基琥珀酰亚胺基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,3.6650g)溶于2.5mL磷酸钠缓冲溶液(pH=5.64±0.02)中,得到第二组分;将第一组分和第二组分通过挤出装置等体积混合挤出,得到医用水凝胶III。
实施例4
<第一医用水凝胶前体(亲核试剂)的制备>
以季戊四醇(5mmol,0.6808g)为起始剂,加入至50mL的反应瓶中,加入10mL吡啶,置于60℃水浴中,抽真空,并通入高纯氮气,反复操作,最后连续抽真空状态下保持2小时,以除去水和氧气。
将温度降至20℃,并持续通入高纯氮气,然后加入环氧丙烷(40mmol,2.3232g,2.7991mL)、甲醇钠(0.05mmol,0.0027g,0.0021mL)引发环氧丙烷开环反应,持续反应2小时;
将温度降至5℃,持续通入高纯氮气,然后加入环氧乙烷单体(80mmol,3.5242g,4.0457mL),持续反应2小时,通过减压蒸馏方式除去吡啶,经纯化后,得到羟基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物,产物质量为4.7201g,产率为72.3%。
取羟基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.7201g,3.6mmol)与亚硫酰二溴(14.4mmol,2.9933g)在二氯甲烷(50mL)和三乙胺(21.6mmol,2.1857g,2.9941mL)中反应得到溴封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.6114g。
将4.6114g的溴封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物与叠氮钠(21.6mmol,1.4042g)在50mL的DMF溶剂中混合,以0.2gPd/C为催化剂,在20mL的乙醇溶剂中催化加氢,获得氨基封端的季戊四醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲核试剂),纯化后称重,质量为4.001g。
<第二医用水凝胶前体(亲电试剂)的制备>
以山梨糖醇(3mmol,0.5465g)为起始剂,加入至50mL的反应瓶中,加入10mL二氧六环,置于60℃水浴中,抽真空,通入高纯氮气,反复操作,最后连续抽真空状态下保持2小时,以除去水和氧气。
将温度降至20℃,并持续通入高纯氮气,加入环氧丙烷(36mmol,2.0909g,2.5192mL)、甲醇钠(0.05mmol,0.0027g,0.0021mL)引发环氧丙烷开环反应,持续反应2小时。
将温度降至5℃,持续通入高纯氮气,然后加入环氧乙烷单体(108mmol,4.7576g,5.4616mL),持续反应2小时,通过减压蒸馏方式除去二氧六环,经纯化后,得到羟基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,产物质量为5.1017g,产率为68.99%。
取羟基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(5.1017g,2.05mmol)与丁二酸酐(18.45mmol,1.8463g)在50mL二氯甲烷和三乙胺(27.675mmol,2.8004g,3.8362mL)中反应,制备得到羧基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物,纯化后称重,质量为4.4189g。
取羧基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(4.4189g)与水溶性碳二亚胺(12.3mmol,2.3579g)、N-羟基琥珀酰亚胺(14.76mmol,1.4625g)于0℃的水溶液中连续搅拌反应30min,得到N-羟基琥珀酰亚胺基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(亲电试剂),纯化后称重,质量为3.9976g。
<医用水凝胶的制备>
称取第一医用水凝胶前体(1mmol,1.3696g)溶于2.5mL磷酸二氢钠与碳酸钠混合的缓冲溶液(pH=9.97±0.02)中,得到第一组分;称取第二医用水凝胶前体溶于2.5mL磷酸钠缓冲溶液(pH=5.64±0.02)中,得到第二组分;将第一组分和第二组分通过挤出装置等体积混合挤出,得到医用水凝胶IV。
标准例1
称取1mmol的3-arm-PEG-NH2,溶于2.5mL的pH=10.0的磷酸钠缓冲溶液中,震荡使其完全溶解。
称取1mmol的4-arm-PEG-NHS,溶于2.5mL的pH=4.0的硼砂缓冲溶液中,震荡使其完全溶解。
将经缓冲溶液溶解后的3-arm-PEG-NH2和4-arm-PEG-NHS通过挤出装置等体积混合挤出,得到医用水凝胶V。
标准例2
称取1mmol的4-arm-PEG-NH2,溶于2.5mL的pH=10.0的磷酸钠缓冲溶液中,震荡使其完全溶解。
称取1mmol的4-arm-PEG-NHS,溶于2.5mL的pH=4.0的硼砂缓冲溶液中,震荡使其完全溶解。
将经缓冲溶液溶解后的4-arm-PEG-NH2和4-arm-PEG-NHS通过挤出装置等体积混合挤出,得到医用水凝胶VI。
测定上述实施例1-4和标准例1-2的成胶时间、溶胀率、破裂强度、剥离拉伸承载强度、开始降解时间以及完全降解时间,结果如下表1所示:
表1
从表1可以看出,本申请实施例1-4制备得到的医用水凝胶I-IV与标准例1-2的医用水凝胶V-VI相比,本申请的水凝胶I-IV同样具有较高的破裂强度和剥离拉伸承载强度,以及较低的溶胀率。因此本申请的水凝胶具有较好的抗破裂强度和粘合力,在组织封闭、创面闭合、防止组织液、血液或脑脊液泄漏方面具有较好的优势。
人工硬脑膜修补实验
取9只健康的大犬作为实验犬,体重均在15公斤以上。随机标号T1,T2,T3,T4,T5,T6,T7,T8,T9。将实验犬麻醉后在其头顶制造2.5cm×2.0cm的硬脑膜缺口,然后进行人工硬脑膜修补手术。所有的实验犬均接受硬脑膜修补术,其中T1、T2、T3为实验组,T4、T5、T6为对照组,T7、T8、T9为空白组。
实验组T1、T2、T3植入人工硬脑膜,采用按照实施例4的方法制备得到的第一组分和第二组分进行动物实验。通过两组分注射器将第一组分和第二组分混合后,喷涂于人工硬脑膜材料周围及表面,30秒后检查其交联情况,待两组分完全交联形成医用水凝胶IV后将伤口封闭缝合。图1为实验组T2接受硬脑膜修补术后,将医用水凝胶IV喷涂于人工硬脑膜周围后的照片。
对照组T4、T5、T6植入人工硬脑膜,采用按照标准例2的方法制备得到的第一组分和第二组分进行动物实验。通过两组分注射器将第一组分和第二组分混合后,喷涂于人工硬脑膜材料周围及表面,30秒后检查其交联情况,待两组分完全交联形成医用水凝胶VI后将伤口封闭缝合。图2为实验组T3接受硬脑膜修补术后,将医用水凝胶VI喷涂于人工硬脑膜周围后的照片。
空白组T7、T8、T9在植入人工硬脑膜后不进行喷涂,直接将伤口封闭缝合。
手术一周后观察,所有的实验犬均保持健康。解剖实验组T1的实验犬和对照组T4的实验犬以及空白组T7的实验犬后发现:空白组T7的实验犬发生了人工硬脑膜侧向移动或与附着位置脱离现象;实验组T1的实验犬在用医用水凝胶IV封闭后未发生人工硬脑膜移动问题,对照组T4的实验犬在用医用水凝胶VI封闭后也未发生人工硬脑膜移动问题。
手术两周后观察,所剩余的实验犬均保持健康。解剖实验组T2的实验犬和对照组T5的实验犬以及空白组T8的实验犬后发现:空白组T8的实验犬发生了人工硬脑膜侧向移动或与附着位置脱离现象,堆积至一侧并发生脑脊液泄漏;实验组T2的实验犬在用医用水凝胶IV封闭后未发生人工硬脑膜移动问题,水凝胶的颜色变淡并可观察到已发生明显的降解。对照组T5的实验犬在用医用水凝胶VI封闭后也未发生人工硬脑膜移动问题。
手术四周后观察,剩余的实验犬均存活,解剖实验组T3的实验犬和对照组T6的实验犬以及空白组T9的实验犬后发现:空白组T9已发生明显的脑脊液外漏问题;实验组T3和对照组T5中的医用水凝胶前体已完全降解,消失不见,且并未发生脑脊液外漏的问题。
另外,在实验组T1~T6中所有的实验犬身上都没有发现神经缺陷,从最初的手术操作开始所有的实验兔均保持健康。
上述结果表明,当采用本申请的医用水凝胶前体制备得到的医用水凝胶,可以有效地应用在人工硬脑膜及邻近组织上,实现组织封闭,防止脑脊液泄露。
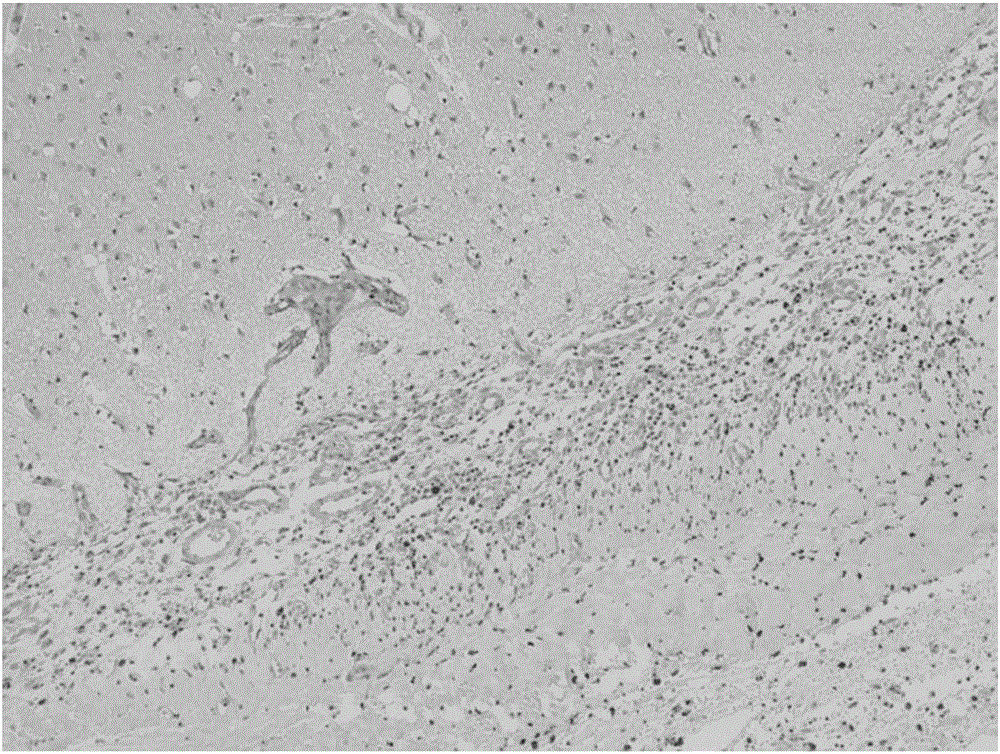
自体硬脑膜修复实验
将实施例2的医用水凝胶II应用于自体硬脑膜修复,两个月后取出修复部位组织做病理切片,采用苏木精-伊红染色后在显微镜下观察。图2是医用水凝胶II应用于自体硬脑膜修复部位的组织切片图,如图2所示,医用水凝胶II已完全降解,并且医用水凝胶II与硬脑膜接触位置未见炎症细胞,硬脑膜组织无变性,无坏死,无纤维化增生。另外,硬脑膜周围其它组织细胞也未见细胞变性和坏死等病理改变。因此,本申请的医用水凝胶具有良好的组织相容性,可完全生物降解。
细胞毒实验
通过浸提液接触培养细胞,以L929为实验细胞,对细胞增殖观察,评价实施例2所制备的医用水凝胶II对体外细胞的毒性作用。
实验的具体过程为:用完全培养基(DMEM培养基+10%胎牛血清(FBS)+1%双抗(青霉素和链霉素的混合液))按照0.1g/mL的比例浸提医用水凝胶,得到水凝胶的浸提液。
实验组:采用完全培养基(DMEM培养基+10%胎牛血清(FBS)+1%双抗(青霉素和链霉素的混合液),添加水凝胶的浸提液,加入细胞悬液,进行细胞培养。
对照组:采用完全培养基(DMEM培养基+10%胎牛血清(FBS)+1%双抗(青霉素和链霉素的混合液))加入细胞悬液,进行细胞培养。对照组中不添加水凝胶的浸提液,其余细胞培养条件与实验组相同。
空白组:采用完全培养基(DMEM培养基+10%胎牛血清(FBS)+1%双抗(青霉素和链霉素的混合液),空白组中不添加水凝胶的浸提液和细胞悬液,于相同的时间放置于与实验组和对照组相同的环境中,以作为实验组和对照组测量吸光度值时的参照。
采用实施例2的医用水凝胶II进行浸提液的配制,浸提温度为(37±1)℃,浸提时间为(24±2)h。采用完全培养基重悬L929细胞,制得浓度为4×104个细胞/mL的细胞悬液。以96孔平板为培养板,其中实验组和对照组为在培养板中加入上述细胞悬液,每孔加入100μL;空白组为在培养板中每孔加入100μL的完全培养基,不加入细胞悬液。
将实验组、对照组和空白组的培养板置于培养箱中预培养24小时(在37℃,5%CO2的条件下)。然后在实验组中加入水凝胶的浸提液,每孔加入100μL,对照组和空白组不加水凝胶的浸提液,之后将各培养板置于培养箱中培养24小时(在37℃,5%CO2条件下)。然后小心吸出孔内的培养液,再向每孔内加入100μL的CCK-8混合液(由90μL的完全培养基+10μL的CCK-8混合制成),再将培养板置于培养箱中孵育2小时。然后用酶标仪测定在450nm处的吸光度。
图3为细胞毒实验中,实验组、对照组分别减去空白组后(即以空白为参照)的吸光度值(OD值)。由图3可以得出,实验组细胞的相对增殖率为RGR=94.92%。
其中实验组细胞的相对增殖率RGR的计算方法为:RGR=(实验组OD值-空白组OD值)/(对照组OD值-空白组OD值)×100%。因此,本申请的医用水凝胶具有良好的安全性,对细胞生长无毒副作用。
负载细胞的生物3D打印实验
将实施例3的氨基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,2.5610g)溶于2.5mL磷酸二氢钠与碳酸钠混合的缓冲溶液(pH=7.97±0.02)中;将实施例3的N-羟基琥珀酰亚胺基封端的山梨糖醇-环氧丙烷-环氧乙烷星形嵌段共聚物(1mmol,3.6650g)溶于2.5mL磷酸钠缓冲溶液(pH=5.64±0.02)中,并加入骨髓间质干细胞,细胞浓度为5×106个/mL。将两组分材料分别灌装在与打印机相连的注射器中,两只注射器在挤出口位置连通混合,然后导入打印机喷头进行负载骨髓间质干细胞的生物3D打印实验。
图4为采用实施例3的医用水凝胶前体经生物3D打印后的支架材料的光学照片,图5为负载骨髓间质干细胞的生物支架的显微镜照片。由图4和图5可以清晰地看到其网格支架结构及骨髓间质干细胞。图6为负载骨髓间质干细胞的生物支架的场发射电子扫描显微镜照片,由图6可以清晰地看到生物支架的支撑的网格结构。因此,通过采用本申请的医用水凝胶前体制备得到的可负载细胞的生物支架,可以为细胞的分裂增殖提供一个良好的生长环境和三维空间,浸润在培养基中可使细胞在生物支架存在的环境中分裂增殖。
- 还没有人留言评论。精彩留言会获得点赞!