野阳合多糖及其制备方法和应用与流程
本发明具体涉及野阳合多糖制备方法、野阳合多糖及其应用,属于植物提取技术领域。
背景技术:
野阳合(HabenariaCiliolarisKranzl),亦称毛葶玉风花(《中国高等植物图鉴》),属于双子叶植物药兰科植物野阳合的块根。原产我国南部,栽培历史悠久,现分布于四川、贵州、安徽、湖北、江西、江苏等省,日本亦有分布。野阳合富含蛋白质、氨基酸、维生素、膳食纤维、有机酸及矿物元素等。野阳合可凉拌、炒食、酱藏、腌渍,也可制作果脯、果汁等。具有镇咳平喘、通经活血、消肿解毒等功效,对治疗便秘、糖尿病有特效。近年来,国内外学者在野阳合资源的生物学特征、栽培技术、贮藏加工、化学成分分析、红色素提取等方面展开探究,研究发现野阳合原为野生,生命力级强,且资源丰富,产量高。为充分有效地开发利用野阳合资源,需系统深入研究其多糖类、多酚类等物质的结构,探明活性机理,且急需一种操作简便,质量可控、提取率高的野阳合活性物质制备方法。
多糖是自然界含量最丰富的生物聚合物,广泛分布在动物、植物、微生物中,是生命体的储能物质和结构物质。现代药理学研究表明,多糖有广泛的生物活性,包括增强生物体免疫调节功能,具有抗肿瘤、抗病毒、抗衰老、降血糖、降血等。
植物多糖广泛分布在植物的根、茎、叶、花、果实及种子中,占植物干重的80%以上。植物资源丰富,植物多糖种类繁多,原料成本低,易于产业化生产。目前,植物多糖在天然产物化学研究中受到越来越大的重视,对植物多糖的研究具有十分重要的应用价值。
技术实现要素:
本发明的目的之一在于提供一种野阳合多糖的制备方法。
本发明的目的之二在于提供四种纯化的野阳合多糖组分。
本发明的目的之三在于提供一种野阳合多糖新的用途。
为实现本发明的第一项发明目的,本发明采用如下技术方案,一种野阳合多糖的制备方法,该方法包括以下步骤:
(1)脱脂:取干燥野阳合粉碎,按料液比1g:20~50mL加入浓度为90wt%的乙醇,在室温下搅拌浸提脱脂,每次12~36h,重复3次,过滤得滤渣;
(2)提取:将上述滤渣干燥处理后,按料液比1g:20~50加入90℃的热水搅拌浸提,每次4~8h,重复3次,分离上清液;
(3)除杂:上清液经浓缩后,用sevage法(氯仿:正丁醇=4∶1)脱蛋白,选用截留量为3500Da的透析袋透析除小分子杂质;
(4)醇沉:收集透析袋内的多糖溶液,减压浓缩,加入浓度为99wt%的乙醇溶液,至多糖浓度大于85%,4℃醇沉过夜,8000r/min离心15min,得粗多糖样品(YP);
(5)DEAE-纤维素52柱层析:将野阳合粗多糖(YP)上样至DEAE-纤维素52柱,依次用纯水、0.1mol/L NaCl、0.3mol/LNaCl和0.5mol/LNaCl洗脱,洗脱速度为:1~2mL/min,每管收集洗脱液3~5mL,苯酚硫酸法检测收集多糖组分,其标准为OD490>0.1,透析除盐,得多糖组分YP1、YP2、YP3、YP4;
(6)TOYOPEARL HW-65柱层析:将经DEAE-纤维素52柱初步分离后的多糖样品YP1、YP2、YP3,分别上样至TOYOPEARL HW-65柱,用纯水洗脱,洗脱速度为:1~2mL/min,每管收集洗脱液3~5mL,苯酚硫酸法检测收集多糖组分,其标准为OD490>0.1,透析除盐,分离得到四个纯化野阳合多糖组分:YP1-1、YP2-1、YP2-2、YP3-1;
(7)冷冻干燥:真空冷冻干燥得到四个纯化野阳合多糖组分。
本发明还提供了四种纯化野阳合多糖组分,是采用上述技术方案以干燥野阳合为原料,经粉碎,90wt%乙醇浸提脱脂,90℃热水浸提多糖,sevage法脱蛋白,透析除杂,乙醇沉淀得粗多糖,经DEAE-纤维素52柱层析和TOYOPEARL HW-65柱层析纯化,真空冷冻干燥分离纯化组分得到的。
结构分析
1、组分检测:采用苯酚-硫酸法检测野阳合粗多糖(YP)总糖含量为96.35%。野阳合粗多糖(YP)水溶液经200~700nm紫外扫描,在260nm和280nm处,没有明显吸收峰,说明该样品中不含核酸和蛋白质,详见图1所示。
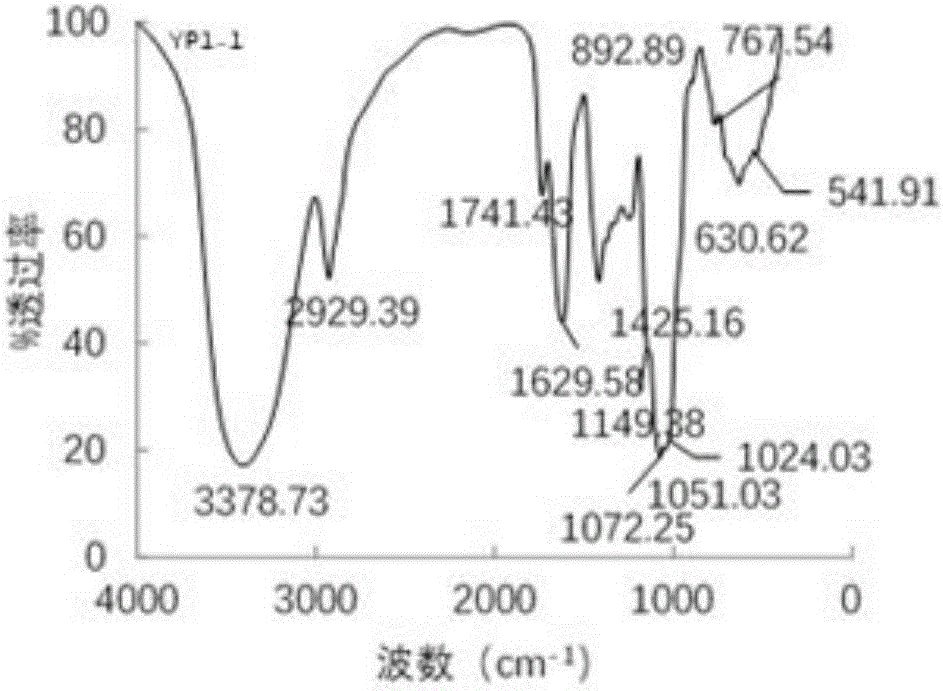
2、FITR:称取3~4mg采用上述技术方案制备的纯化野阳合多糖组分YP1-1(以YP1-1为例,其它如YP2-1、YP2-2、YP3-1同理),用KBr压片,Nicolet 6700型红外光谱仪进行红外扫描,详见图2所示。
3、分子量:采用高效凝胶渗透色谱法(HPGPC),测定野阳合多糖的分子量分布,详见图3a、图3b、图3c、图3d所示。
4、单糖组成:称取野阳合多糖YP1-1(以YP1-1为例,其它如YP2-1、YP2-2、YP3-1同理)10~15mg,加入2M三氟乙酸(TFA)4~5mL,封管,110℃水解6~8h,冷却至室温,取管内溶液减压蒸干(低于40℃),反复加入(重复操作4~5次)甲醇,减压蒸干,以完全除去TFA。用超纯水溶解水解样品并定容至100mL容量瓶,采用高效液相色谱-蒸发光散射检测(HPLC-ELSD)法测定,详见表1所示。
表1野阳合多糖的单糖组成a
a数据为样品单糖组成的摩尔比
结构分析表明四个纯化野阳合多糖组分YP1-1、YP2-1、YP2-2、YP3-1分子量分别为:4.29×106Da、3.40×106Da、2.10×106Da和4.09×106Da、;单糖组成结果见表1所示。
本发明还提供了野阳合多糖的新用途。野阳合多糖体外结合胆汁酸能力较强,作为降胆固醇的活性物质。
本发明具有如下优点:
本发明提供了野阳合多糖的制备方法,该方法对设备要求低、成本低,可用于工业生产,利用本发明制备野阳合多糖,不影响多糖的天然结构和活性。
附图说明
图1为实施例野阳合粗多糖(YP)在200~700nm的紫外扫描图谱。
图2a为实施例纯化野阳合多糖组分YP1-1的红外光谱图。
图2b为实施例纯化野阳合多糖组分YP2-1的红外光谱图。
图2c为实施例纯化野阳合多糖组分YP2-2的红外光谱图。
图2d为实施例纯化野阳合多糖组分YP3-1的红外光谱图。
图3a为实施例纯化野阳合多糖组分YP1-1的分子量分布图谱。
图3b为实施例纯化野阳合多糖组分YP2-1的分子量分布图谱。
图3c为实施例纯化野阳合多糖组分YP2-2的分子量分布图谱。
图3d为实施例纯化野阳合多糖组分YP3-1的分子量分布图谱。
图4为实施例野阳合粗多糖(YP)经DEAE-纤维素52柱层析分离的洗脱曲线。
具体实施方式
下面通过实施例对本发明进行具体的描述,但本发明不局限于此实施例,不能理解为对本发明保护范围的限制,实际操作时,技术人员可依据本发明的内容作出一定程度的非本质的改进与调整。
实施例1:野阳合多糖的制备:
脱脂:称取干燥野阳合100g,粉碎,按料液比1g:20~50mL加入2~5L浓度为90wt%的乙醇,在室温下搅拌浸提脱脂,每次12~36h,重复3次,过滤分离得滤渣73.91g;
(2)提取:将干燥后的滤渣,共73.91g,按料液比1g:20~50mL加入90℃的热水3.7L浸提多糖,每次4~8h,重复3次,过滤分离得滤液;
(3)除杂:滤液经减压浓缩后,用sevage法(氯仿:正丁醇=4∶1)脱蛋白,重复操作6次,选用截留量为3500Da的透析袋透析除小分子杂质;
(4)醇沉:收集透析袋内的多糖溶液,减压浓缩,加入浓度为99wt%的乙醇溶液,至多糖浓度大于85wt%,4℃醇沉过夜,8000r/min离心15min,得粗多糖样品YP,共计1.42g;
(5)DEAE-纤维素52柱层析:取野阳合粗多糖1g,溶解于10mL超纯水,上样至DEAE-纤维素52柱,依次用纯水、0.1mol/L NaCl、0.3mol/LNaCl和0.5mol/L NaCl洗脱,洗脱速度为:1~2mL/min,每管收集洗脱液3~5mL,苯酚硫酸法检测收集OD490>0.1的多糖组分,透析除盐,得初步多糖组分YP1、YP2、YP3、YP4;野阳合粗多糖(YP)经DEAE-纤维素52柱层析分离的洗脱曲线如图4所示。
(6)TOYOPEARL HW-65柱层析:称取0.5g多糖组分YP1(以YP1为例,其它如YP2、YP3同理)溶于4~6mL超纯水,上样至TOYOPEARL HW-65柱,用纯水洗脱,洗脱速度为:1~2mL/min,每管收集洗脱液3~5mL,苯酚硫酸法检测并收集OD490>0.1的多糖组分,透析除盐,分离得到四个纯化野阳合多糖组分:YP1-1、YP2-1、YP2-2、YP3-1;
(7)真空冷冻干燥:真空冷冻干燥得到四个纯化野阳合多糖组分。
下面是对四个纯化野阳合多糖组分进行结构分析的实施例:
实施例2:组分检测
将实施例1中制得的野阳合粗多糖(YP),用苯酚-硫酸法检测,其总糖含量为97.27%。野阳合粗多糖(YP)水溶液进行200~700nm紫外扫描,结果见图1,在260nm和280nm处,没有明显吸收峰,说明该样品中不含核酸和蛋白质。
实施例3:FITR
取3~4mg实施例1中制得的四个纯化野阳合多糖组分:YP1-1、YP2-1、YP2-2、YP3-1,分别用KBr压片,Nicolet 6700型红外光谱仪在4000~500cm-1范围内进行红外扫描。如图2a、图2b、图2c和图2d所示。
四个野阳合多糖组分YP1-1、YP2-1、YP2-2及YP3-1分别在3378.73cm-1、3382.58cm-1、3394.16cm-1和3386.44cm-1处出现宽而强的吸收带,它们是分子内或分子间氢键的O-H伸缩振动的结果;分别在2929.39cm-1、2927.46cm-1、2925.6cm-1和2939.03cm-1处出现的吸收带是糖类C-H伸缩振动的结果;分别在1741.43cm-1、1727.93cm-1、1729.86cm-1和1722.15cm-1处出现的强吸收带是多糖样品中乙酰基、酯基或羧基中C=O非对称伸缩振动的结果;分别在1629.59cm-1、1608.37cm-1、1612.22cm-1和1637.30cm-1处出现的吸收带,表明多糖含有一定量的结合水;分别在1425.16cm-1、1413.59cm-1、1413.59cm-1和1400.09cm-1处出现的吸收带是糖类C-H变角振动的结果;野阳合多糖YP1-1、YP2-1及YP2-2分别在1330.66cm-1、1326.81cm-1和1332.59cm-1处的吸收带是羰基O-H弯曲振动的结果;四个野阳合多糖组分YP1-1、YP2-1、YP2-2及YP3-1分别在1238.10cm-1、1241.95cm-1、12138.10cm-1和1274.74cm-1处出现吸收带是乙酰基(C-O)伸缩振动的结果;野阳合多糖YP1-1、YP2-1及YP2-2在1100~1000cm-1范围内均出现三个较强的吸收带,表明多糖组分含有吡喃糖,野阳合多糖YP3-1在1100~1000cm-1范围内均出现两个较强的吸收带,表明多糖组分含呋喃糖有吡喃糖;野阳合多糖YP1-1在892.89cm-1处出现的吸收带,表明该多糖是以β-型糖苷键连接;野阳合多糖YP2-1在894.82cm-1和829.25cm-1处出现的吸收带,表明该多糖是以β-型和α-型糖苷键连接;野阳合多糖YP2-2在890.97cm-1和829.25cm-1处出现的吸收带,表明该多糖是以β-型和α-型糖苷键连接;野阳合多糖YP1-1在889.04cm-1处出现的吸收带,表明该多糖是以β-型糖苷键连接。
实施例4:单糖组成
实施例1制得的野阳合多糖YP1-1(以YP1-1为例,其它如YP2-1、YP2-2、YP3-1同理)10mg,加入2M三氟乙酸45,封管,110℃水解6h,冷却至室温,取试管内溶液减压蒸干(低于40℃),反复加入(重复操作4~5次)甲醇,减压蒸干,以完全除去TFA。用超纯水溶解定容至100mL容量瓶,上样HPLC-ELSD测定。
HPLC条件:柱子,TSKgel Amide-80(5μm,4.6×250nm);流速,1.0mL/min;柱温,80℃;进样体积,20μL;流动相A(超纯水):流动相B(乙腈)=15∶85。如表1所示。野阳合多糖YP1-1的单糖组成主要为鼠李糖、阿拉伯糖、葡萄糖和半乳糖,其物质的量比为2.2∶13.1∶10∶5.7;野阳合多糖YP2-1的单糖组成主要为鼠李糖、阿拉伯糖、甘露糖、葡萄糖和半乳糖,其物质的量比为3.5∶8.5∶1.8∶9.4∶9.3;野阳合多糖YP2-2的单糖组成主要为鼠李糖、阿拉伯糖、甘露糖、葡萄糖和半乳糖,其物质的量比为7.2∶9.2∶1.2∶10∶6.5;野阳合多糖YP3-1的单糖组成主要为鼠李糖、阿拉伯糖、甘露糖、葡萄糖和半乳糖,其物质的量比为9.2∶1.9∶1∶9.4∶9.3。
实施例5:分子量测定
采用高效凝胶渗透色谱法(HPGPC),测定野阳合多糖的分子量分布。
HPLC条件:柱子,TSKgel G5000PWXL(10μm,7.8×300nm);流速,0.6mL/min;柱温,35℃;进样体积,20μl;流动相,超纯水。
实施例6:胆汁酸结合实验
将0.5mL多糖样品溶液(5~25mg/mL)和0.25mL 0.01N HCl在37℃下搅拌1h。向混合物中加入1mL胆汁酸混合溶液并搅拌混匀,随后加入2mL猪胰酶溶液提供消化用酶。将混合物在37℃下反应1h。用5倍体积的乙醇沉淀野阳合多糖,并离心(5000×g,15~20min)。通过BECKMAN COULTER AU480全自动生化仪测定上清液中胆汁酸的最终含量,确定被多糖结合绑定的胆汁酸含量。辛伐他汀为阳性对照。计算野阳合多糖相比于辛伐他汀(定义为100)结合胆汁酸的能力。所有分析均进行3次。野阳合粗多糖和四种纯化的野阳合多糖组分对胆汁酸的结合能力如表2所示。
表2野阳合多糖相对辛伐他汀对胆汁酸的结合能力
辛伐他汀能增加胆汁酸分泌,并有效降低低密度脂蛋白胆固醇(LDL-C)吸收,在本实验中作为阳性对照,其对胆汁酸的结合能力定义为100。由表2可见,野阳合粗多糖和四种纯化的野阳合多糖组分对胆汁酸的结合能力均高于97,有强胆汁酸结合能力。
- 还没有人留言评论。精彩留言会获得点赞!