一种合成天然产物丹酚酸A甲酯的方法与流程
本发明属于化工领域。涉及一类具有抗氧化作用、抗血栓形成、抗血小板活性、抗糖尿病、保护急性肝脏损伤、抗肝纤维化、和预防肿瘤、抑制肿瘤细胞增长等作用的天然产物丹酚酸A甲酯的合成新方法。其关键策略及步骤包括利用易于去除的叔丁基二甲基硅基保护基作为酚羟基的保护基以避免副反应,利用Sharpless不对称双羟化反应再用乙基硅烷还原构建C-8'位的羟基手性中心,采用Horner-Wadsworth-Emmons反应来构建丹酚酸A甲酯中C7-9位的α,β-不饱和酯结构。
技术背景
天然产物丹酚酸A甲酯(如图1)是由陈政雄等人于20世纪80年代,在多种丹参水溶性成分进行系统研究时,运用多种色谱分离提纯的技术从丹参中的水溶性成分中分离到十几种丹酚酸类化合物,其中包括丹酚酸A甲酯。之后的药理学活性研究表明,丹酚酸A甲酯具有显著的抗氧化、抗血栓形成、抗血小板活性、抗糖尿病、保护急性肝脏损伤、抗肝纤维化和预防肿瘤等作用。其良好的药理活性和生物活性,吸引了众多化学合成家、生物医学家及药理学家的极大兴趣。
对于丹酚酸A和丹酚酸A甲酯的合成报道目前还没有,其原因可能是由于裸露的酚羟基容易引起很多副反应。因此,本发明针对酚羟基的保护基选用易脱除的叔丁基二甲基硅基保护基,筛选出合适的脱除方法,最终成功制备丹酚酸A甲酯。
技术实现要素:
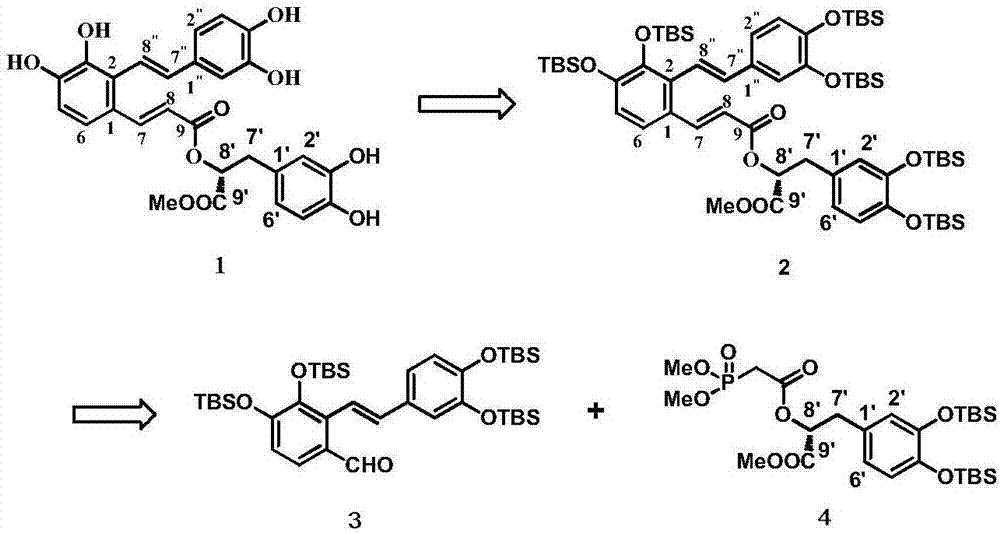
本发明提供的丹酚酸A甲酯的合成方法旨在实现丹酚酸A甲酯的人工合成,因此设计以廉价易得的4-甲基儿茶酚,邻香草醛,咖啡酸等为基本原料,采用汇聚式合成策略实现。其逆合成设计如图2所示:终产物丹酚酸A甲酯1的合成最后一步设计通过化合物2脱除羟基脱保护基得到,2当中的C7-9的α,β-不饱和酯结构的合成设计由片段3和片段4经Horner-Wadsworth-Emmons反应合成得到。其中片段3的合成由4-甲基儿茶酚经过羟基保护,溴代,Wittig反应等制得,片段4的合成经Sharpless双羟基化,三乙基硅烷还原,酯化反应等制得,最后将制备的片段3和片段4经Horner-Wadsworth-Emmons反应,脱保护基等过程得到丹酚酸A甲酯。
本发明技术方案如下:
一种合成天然产物丹酚酸A甲酯的方法,丹酚酸A甲酯即式Ⅰ化合物:由式Ⅱ化合物
通过三乙胺氢氟酸盐的条件脱除叔丁基二甲基硅基保护基后得到式Ⅰ化合物;
式Ⅱ化合物获得:式Ⅲ化合物,Ⅳ化合物
通过Horner-Wadsworth-Emmons反应构建C7-9的α,β-不饱和酯结构得到式Ⅱ化合物;
式Ⅲ化合物获得:式Ⅴ,Ⅵ化合物
通过Wittig反应构建C7”-8”不饱和碳链得到式Ⅲ化合物;
式Ⅳ化合物获得:包括式Ⅶ,Ⅷ化合物
通过酯化反应来获得式Ⅳ化合物;
式Ⅶ化合物获得:式Ⅸ化合物
通过Sharplesss双羟化的条件对C7'-8'进行双羟基化后,然后在三氟乙酸与三乙基硅烷的条件下来构建C8'位的单一羟基手性中心,得到式Ⅶ化合物。
通过以邻香草醛为原料来获得Ⅴ化合物。
通过以4-甲基儿茶酚为原料来获得式Ⅵ化合物。
通过以咖啡酸为原料来获得式Ⅸ化合物。
所述的方法,步骤具体为:
a.首先合成磷鎓盐7,可以由初始原料4经两步合成得到:
(1)式9化合物的合成
在0℃时,将式8化合物4-甲基儿茶酚(5.0g,40.28mmol)溶于N,N-二甲基甲酰胺(DMF)中,氮气保护下,加入咪唑(13.7g,201.39mmol)和叔丁基二甲基氯硅烷(TBSCl)(18.2g,120.83mmol),然后升至常温,搅拌过夜;点板监测反应至完全结束,之后,用冰水淬灭反应,然后用二氯甲烷萃取(3×150mL),合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂后,经色谱柱分离得式9化合物14.20g:
(2)式7化合物的合成
化合物9(14.2g,40.27mmol)溶于四氯化碳(80mL)中,氮气保护下加入N-溴代丁二酰亚胺(NBS)(7.88g,44.29mmol)和偶氮二异腈(0.979g,6.04mmol),回流1小时后,点板监测反应至完全结束,加冰水淬灭反应,然后用二氯甲烷萃取(3×100mL),合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂后,加80mL乙腈溶解,再加入三苯基膦(15.84g,60.41mmol),回流45分钟后,将溶剂减压浓缩,再加甲苯洗涤,过滤后得式7化合物为白色粉末(15.95g);
b.接下来合成片段醛5,可以由初始原料6经五步合成得到;
(3)式10化合物的合成
在0℃时,氮气保护下将邻香草醛6(10.01g,65.72mmol)和催化量的4-二甲氨基吡啶(0.80g,7.89mmol)溶解在二异丙基乙基胺(32.86mL),加入醋酸酐(8.07mL,85.44mmol),然后升至常温,搅拌过夜,点板监测至反应完全结束;加2N的HCl溶液100mL,再加二氯甲烷(3×100mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;用无水乙醇进行重结晶,得式10化合物为黄色晶体10.85g;
(4)式11化合物的合成
在0℃时,溴化钾(21.47g,180.48mmol)溶于水133mL中,缓慢加入溴素(3.73mL,72.64mmol),之后向深红色水溶液中加入化合物10(10.85g,55.88mmol),形成的桔红色水溶液在常温下搅拌过夜,点板监测至反应完全结束;过滤,乙酸乙酯洗涤,然后用乙酸乙酯和正己烷进行重结晶,得式11化合物为黄色晶体11.44g;
(5)式12化合物的合成
化合物11(11.44g,41.89mmol)溶于甲醇水溶液(84mL),加入NaHCO3(4.93g,58.64mmol),形成的黄色浑浊溶液在常温搅拌2小时,点板监测至反应完全结束,用盐酸酸化,加二氯甲烷(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;用乙酸乙酯和正己烷进行重结晶,得式12化合物为黄色晶体8.23g;(6)式13化合物的合成
在0℃时,氮气保护下,化合物12(8.23g,35.62mmol)溶于干燥的二氯甲烷(120mL)中,将三溴化硼(13.47mL,142.48mmol)溶于也干燥的二氯甲烷(60mL)中,再将此混合液缓慢加入至反应液当中,温度升至常温,搅拌2小时,点板监测至反应完全结束;加饱和NaHCO3溶液终止反应,再加二氯甲烷(3×100mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离后得式13化合物为黄色固体7.34g;
(7)式5化合物的合成
在0℃时,化合物13(7.34g,33.82mmol)溶于干燥的N,N-二甲基甲酰胺(307mL),氮气保护下加入三乙胺(16.50mL,118.37mmol),4-二甲氨基吡啶(0.826g,6.76mmol)和叔丁基二甲基氯硅烷(TBSCl)(15.29g,101.46mmol),温度升至常温,搅拌过夜,点板监测至反应完全结束;加水终止反应,再加二氯甲烷(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离后可得式5化合物为黄色油状产物13.86g;
c.再接下来,醛3的合成可通过以下两步反应合成得到:
(8)式14化合物的合成
在-78℃时,将磷鎓盐7(19.47g,28.06mmol)溶于干燥的四氢呋喃(100mL),氮气保护下加入正丁基锂(25.81mmol),搅拌30分钟后,将醛5(5.0g,11.22mmol)溶于40mL干燥的四氢呋喃中,再将此混合液在在-78℃下缓慢加入至反应瓶中,并在该温度下搅拌2小时,点板监测至反应完全反应结束;加饱和氯化铵终止反应,再加乙酸乙酯(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离后得式14化合物无色油状产物6.56g;
(9)式3化合物的合成
在-78℃时,将化合物14(6.56g,8.41mmol)溶于干燥的四氢呋喃(40mL)中,氮气保护下加入正丁基锂(16.82mmol),搅拌30分钟后,将N,N-二甲基甲酰胺(6.48mL,84.08mmol)溶于干燥的四氢呋喃(10mL),将此混合液在-78℃下缓慢加入至反应瓶中,该温度下搅拌3小时,点板监测至反应完全结束,加饱和氯化铵终止反应,再加乙酸乙酯(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离后得式3化合物5.21g;
d.再接下来,手性磷酸酯4的合成可通过以下几步反应合成得到:
(10)式16化合物的合成
氮气保护下,将咖啡酸15(2.0g,11.10mmol)溶于140mL甲醇中,然后加入浓硫酸4mL,加热至70℃,回流反应1小时,点板监测至反应完全结束;然后冷却至室温,加入饱和碳酸氢钠水溶液中和反应,再加二氯甲烷(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂,得粗产物式16化合物为灰色固体2.15g;
(11)式17化合物的合成
在0℃时,氮气保护下,将式16化合物(2.15g,11.07mmol)溶解在60mL干燥的N,N-二甲基甲酰胺(DMF)中,加入咪唑(5.28g,77.51mmol)和叔丁基二甲基氯硅烷(TBSCl)(5.0g,33.22mmol),温度升至常温后,然后在该温度下反应6小时,点板监测至反应完全结束;加水淬灭反应,再加乙酸乙酯(3×60mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离纯化后得式17化合物为白色固体4.74g;
(12)式18化合物的合成
氮气保护下,将AD-mix-α(15.25g,15.19mmol)和甲基磺酰胺(20mmol)溶解在80mL叔丁醇中,剧烈搅拌,直至两相溶液都变成透明澄清状态;然后将该混合物转移至0℃,将化合物17(4.74g,10.85mmol)溶解在40mL的叔丁醇里,缓慢加入至上述透明溶液中,温度升至常温,该温度下搅拌反应28小时,点板监测至完全结束;之后,再次将该混合溶液转移至0℃,加入亚硫酸钠(5g),温度升至常温后,再继续搅拌1小时,再加乙酸乙酯(3×60mL)萃取,合并有机相,加2N的氢氧化钾水溶液洗涤,之后再用饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;所得的粗产物经硅胶柱层析分离纯化后得化合物18为白色固体4.46g;
(13)式19化合物的合成
在0℃时,氮气保护下,将式18化合物(4.46g,9.77mmol)溶解在40mL干燥的二氯甲烷中,加入三乙基硅烷(48.83mmol)和三氟乙酸(97.7mmol),0℃下搅拌反应10小时,点板监测至反应完全结束;加饱和碳酸氢钠水溶液淬灭反应,再加乙酸乙酯(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离纯化后得无色油状化合物19为3.44g;
(14)式4化合物的合成
在0℃时,氮气保护下,将式19化合物(3.44g,7.81mmol)和三甲基膦酰基乙酸(12.49mmol)溶解在40mL干燥的二氯甲烷中,加入二环己基碳二亚胺(DCC)(12.49mmol)和4-二甲氨基吡啶(DMAP)(0.19g,1.56mmol),常温下搅拌1小时,点板监测至反应完全结束;加水溶液淬灭反应,再加二氯甲烷(3×20mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离纯化后得到白色半固体化合物4为4.15g;
e.(15)式2化合物的合成
在-78℃时,氮气保护下,将化合物4(415mg,0.703mmol)溶解在5mL干燥的四氢呋喃溶液中,加入正丁基锂(0.29mmol),该温度下搅拌30分钟;然后将化合物3(427mg,0.59mmol)溶解在2mL干燥的四氢呋喃中,逐滴加入到-78℃低温混合物中,该温度下搅拌2小时,点板监测至反应完全结束;加饱和氯化铵水溶液淬灭反应,再加乙酸乙酯(3×15mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离纯化后得式2化合物为黄色固体0.56g;
(16)丹酚酸A甲酯1的合成
在0℃时,将化合物2(500mg,0.42mmol)溶于4mL干燥的吡啶中,氮气保护下加入三乙胺氢氟酸盐(5.02mmol),温度升高至常温,搅拌0.5小时,点板监测至反应完全结束;加冰水终止反应,再加二氯甲烷(3×20mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂;粗产物经硅胶柱层析分离后得终产物1为黄色固体0.19g。
本发明的创新点如下:一是丹酚酸A甲酯目前世界上尚未有人工合成的报道,本发明涉及到的合成路线提供了一条首次人工合成丹酚酸A甲酯的工艺路线;二是针对丹酚酸A甲酯的酚羟基易在后续的脱除中造成很大困难的特点,本发明选用易脱除的叔丁基二甲基硅基保护基,并在后期筛选出合适的脱除方法,最终成功制备丹酚酸A甲酯,这也为丹酚酸A系列化合物裸露酚羟基的人工合成提供了非常好的借鉴意义。三是整个合成路线的设计独特新颖,其反应过程原料易得,条件温和,速率快,副反应较少,操作简便,稳定性和重复性好。
说明书附图
图1丹酚酸A甲酯的化学结构
图2丹酚酸A甲酯的逆合成设计
图3丹酚酸A甲酯中片段3的合成路线
图4丹酚酸A甲酯的合成路线
具体实施方式
本设计所涉及到的合成路线如图3、图4所示。
为了使本发明更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
实施例1
1、首先合成磷鎓盐7,可以由初始原料4经两步合成得到。
(1)式9化合物的合成
在0℃时,将4-甲基儿茶酚8(5.0g,40.28mmol)溶于N,N-二甲基甲酰胺(DMF)中,氮气保护下,加入咪唑(13.7g,201.39mmol)和叔丁基二甲基氯硅烷(TBSCl)(18.2g,120.83mmol),然后升至常温,搅拌过夜。点板监测反应至完全结束,之后,用冰水淬灭反应,然后用二氯甲烷萃取(3×150mL),合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂后,经色谱柱分离得式9化合物14.20g,收率99%。
(2)式7化合物的合成
化合物9(14.2g,40.27mmol)溶于四氯化碳(80mL)中,氮气保护下加入N-溴代丁二酰亚胺(NBS)(7.88g,44.29mmol)和偶氮二异腈(0.979g,6.04mmol),回流1小时后,点板监测反应至完全结束,加冰水淬灭反应,然后用二氯甲烷萃取(3×100mL),合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂后,加80mL乙腈溶解,再加入三苯基膦(15.84g,60.41mmol),回流45分钟后,将溶剂减压浓缩,再加甲苯洗涤,过滤后得式7化合物为白色粉末(15.95g),两步反应总产率70%。
2、接下来合成片段醛5,可以由初始原料6经五步合成得到。
(3)式10化合物的合成
在0℃时,氮气保护下将邻香草醛6(10.01g,65.72mmol)和催化量的4-二甲氨基吡啶(0.80g,7.89mmol)溶解在二异丙基乙基胺(32.86mL),加入醋酸酐(8.07mL,85.44mmol),然后升至常温,搅拌过夜,点板监测至反应完全结束。加2N的HCl溶液100mL,再加二氯甲烷(3×100mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。用无水乙醇进行重结晶,得式10化合物为黄色晶体10.85g,产率85%。
(4)式11化合物的合成
在0℃时,溴化钾(21.47g,180.48mmol)溶于水133mL中,缓慢加入溴素(3.73mL,72.64mmol),之后向深红色水溶液中加入化合物10(10.85g,55.88mmol),形成的桔红色水溶液在常温下搅拌过夜,点板监测至反应完全结束。过滤,乙酸乙酯洗涤,然后用乙酸乙酯和正己烷进行重结晶,得式11化合物为黄色晶体11.44g,产率75%。
(5)式12化合物的合成
化合物11(11.44g,41.89mmol)溶于甲醇水溶液(84mL),加入NaHCO3(4.93g,58.64mmol),形成的黄色浑浊溶液在常温搅拌2小时,点板监测至反应完全结束,用盐酸酸化,加二氯甲烷(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。用乙酸乙酯和正己烷进行重结晶,得式12化合物为黄色晶体8.23g,产率85%。
(6)式13化合物的合成
在0℃时,氮气保护下,化合物12(8.23g,35.62mmol)溶于干燥的二氯甲烷(120mL)中,将三溴化硼(13.47mL,142.48mmol)溶于也干燥的二氯甲烷(60mL)中,再将此混合液缓慢加入至反应液当中,温度升至常温,搅拌2小时,点板监测至反应完全结束。加饱和NaHCO3溶液终止反应,再加二氯甲烷(3×100mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离后得式13化合物为黄色固体7.34g,产率95%。
(7)式5化合物的合成
在0℃时,化合物13(7.34g,33.82mmol)溶于干燥的N,N-二甲基甲酰胺(307mL),氮气保护下加入三乙胺(16.50mL,118.37mmol),4-二甲氨基吡啶(0.826g,6.76mmol)和叔丁基二甲基氯硅烷(TBSCl)(15.29g,101.46mmol),温度升至常温,搅拌过夜,点板监测至反应完全结束。加水终止反应,再加二氯甲烷(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离后可得式5化合物为黄色油状产物13.86g,产率92%。
3、再接下来,醛3的合成可通过以下两步反应合成得到:
(8)式14化合物的合成
在-78℃时,将磷鎓盐7(19.47g,28.06mmol)溶于干燥的四氢呋喃(100mL),氮气保护下加入正丁基锂(25.81mmol),搅拌30分钟后,将醛5(5.0g,11.22mmol)溶于40mL干燥的四氢呋喃中,再将此混合液在在-78℃下缓慢加入至反应瓶中,并在该温度下搅拌2小时,点板监测至反应完全反应结束。加饱和氯化铵终止反应,再加乙酸乙酯(3×80mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离后得式14化合物无色油状产物6.56g,产率75%。
(9)式3化合物的合成
在-78℃时,将化合物14(6.56g,8.41mmol)溶于干燥的四氢呋喃(40mL)中,氮气保护下加入正丁基锂(16.82mmol),搅拌30分钟后,将N,N-二甲基甲酰胺(6.48mL,84.08mmol)溶于干燥的四氢呋喃(10mL),将此混合液在-78℃下缓慢加入至反应瓶中,该温度下搅拌3小时,点板监测至反应完全结束,加饱和氯化铵终止反应,再加乙酸乙酯(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离后得式3化合物5.21g,产率85%。
4、再接下来,手性磷酸酯4的合成可通过以下几步反应合成得到:
(10)式16化合物的合成
氮气保护下,将咖啡酸15(2.0g,11.10mmol)溶于140mL甲醇中,然后加入浓硫酸4mL,加热至70℃,回流反应1小时,点板监测至反应完全结束。然后冷却至室温,加入饱和碳酸氢钠水溶液中和反应,再加二氯甲烷(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂,得粗产物式16化合物为灰色固体2.15g,产率100%。
(11)式17化合物的合成
在0℃时,氮气保护下,将式16化合物(2.15g,11.07mmol)溶解在60mL干燥的N,N-二甲基甲酰胺(DMF)中,加入咪唑(5.28g,77.51mmol)和叔丁基二甲基氯硅烷(TBSCl)(5.0g,33.22mmol),温度升至常温后,然后在该温度下反应6小时,点板监测至反应完全结束。加水淬灭反应,再加乙酸乙酯(3×60mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离纯化后得式17化合物为白色固体4.74g,产率98%。
(12)式18化合物的合成
氮气保护下,将AD-mix-α(15.25g,15.19mmol)和甲基磺酰胺(20mmol)溶解在80mL叔丁醇中,剧烈搅拌,直至两相溶液都变成透明澄清状态。然后将该混合物转移至0℃,将化合物17(4.74g,10.85mmol)溶解在40mL的叔丁醇里,缓慢加入至上述透明溶液中,温度升至常温,该温度下搅拌反应28小时,点板监测至完全结束。之后,再次将该混合溶液转移至0℃,加入亚硫酸钠(5g),温度升至常温后,再继续搅拌1小时,再加乙酸乙酯(3×60mL)萃取,合并有机相,加2N的氢氧化钾水溶液洗涤,之后再用饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。所得的粗产物经硅胶柱层析分离纯化后得化合物18为白色固体4.46g,产率90%,ee值90%。
(13)式19化合物的合成
在0℃时,氮气保护下,将式18化合物(4.46g,9.77mmol)溶解在40mL干燥的二氯甲烷中,加入三乙基硅烷(48.83mmol)和三氟乙酸(97.7mmol),0℃下搅拌反应10小时,点板监测至反应完全结束。加饱和碳酸氢钠水溶液淬灭反应,再加乙酸乙酯(3×50mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离纯化后得无色油状化合物19为3.44g,产率80%。
(14)式4化合物的合成
在0℃时,氮气保护下,将式19化合物(3.44g,7.81mmol)和三甲基膦酰基乙酸(12.49mmol)溶解在40mL干燥的二氯甲烷中,加入二环己基碳二亚胺(DCC)(12.49mmol)和4-二甲氨基吡啶(DMAP)(0.19g,1.56mmol),常温下搅拌1小时,点板监测至反应完全结束。加水溶液淬灭反应,再加二氯甲烷(3×20mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离纯化后得到白色半固体化合物4为4.15g,产率90%。
关键手性磷酸酯中间体4拿到之后,接下来就是通过以下的两步反应来合成最终所需的天然产物Salvianolic Acid A甲酯(1):
(15)式2化合物的合成
在-78℃时,氮气保护下,将化合物4(415mg,0.703mmol)溶解在5mL干燥的四氢呋喃溶液中,加入正丁基锂(0.29mmol),该温度下搅拌30分钟。然后将化合物3(427mg,0.59mmol)溶解在2mL干燥的四氢呋喃中,逐滴加入到-78℃低温混合物中,该温度下搅拌2小时,点板监测至反应完全结束。加饱和氯化铵水溶液淬灭反应,再加乙酸乙酯(3×15mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离纯化后得式2化合物为黄色固体0.56g,产率80%。
(16)丹酚酸A甲酯1的合成
在0℃时,将化合物2(500mg,0.42mmol)溶于4mL干燥的吡啶中,氮气保护下加入三乙胺氢氟酸盐(5.02mmol),温度升高至常温,搅拌0.5小时,点板监测至反应完全结束。加冰水终止反应,再加二氯甲烷(3×20mL)萃取,合并有机相,加饱和NaCl溶液洗涤,无水硫酸钠干燥,过滤,减压蒸出溶剂。粗产物经硅胶柱层析分离后得终产物1为黄色固体0.19g,产率90%。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!