一种茂基双核椅状钴氮配合物及其制备方法和应用与流程
本发明涉及一种钴氮配合物,特别涉及一种新型茂基双核椅状钴氮配合物极其制备方法和其作为催化剂在聚酯合成中的应用,属于新型催化材料合成领域。
背景技术:
近年来,随着后过渡金属配合物在烯烃聚合和原子转移自由基聚合中的成功应用,对后过渡金属镍,钴配合物的合成及其催化性能研究已引起了人们极大的兴趣。目前,后过渡金属钴,钴配合物的研究重点之一是开发高活性的、具有一定稳定性的聚合催化剂。已有的大量研究结果表明:金属有机化合物的催化性能并不完全取决于中心金属原子,还取决于中心金属的配位环境即配体的类型、电子和空间结构,因此,设计合成新的配体,使中心金属具有合适于聚合反应所需的电荷效应和空间结构就成为钴,钴系催化剂开发的必经之路。
Zargarina等人报道了一系列含磷配体的芴基镍,钴配合物的合成及其催化性能的研究,例如芴基钴,钴化合物(Ind)M(Ni,Co)(PR3)X与MAO作用可用于硅烷的聚合、烯烃的聚合及炔烃的聚合,其阳离子化物可用于烯烃二聚,苯乙烯的齐聚等,表现出茂基镍,钴配合物丰富的反应性能。但是这类配合物的稳定性和活性还有待进一步的改善。
技术实现要素:
针对现有技术中存在的缺陷,本发明的一个目的是在于提供一种具有稳定茂基双核椅状结构的钴氮配合物。
本发明的另一个目的是在于提供一种操作简单、反应条件温和的制备所述茂基双核椅状钴氮配合物的方法,该方法制备的产品晶体相均匀,纯度和产率高。
本发明的第三个目的是在于提供一种所述茂基双核椅状钴氮配合物作为催化剂的应用,将其应用于催化酯化反应中体现出较高的催化反应活性。
为了实现上述技术目的,本发明提供了一种茂基双核椅状钴氮配合物,该配合物具有式1化学表达式:
{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O
式1
其中,
C12H8N2的结构式为:
C7H8(COO)2的结构式为:
本发明的茂基双核椅状钴氮配合物的分子结构式如下式2:
优选的方案,茂基双核椅状钴氮配合物属单斜晶系,空间群为P2(1)/c;晶胞参数:α=90°,β=98.85(3)°,γ=90°,Mr=455.73,Dc=1.498g/cm3,Z=8,μ(MoKα)=1.12mm-1,F(000)=1396。
所述的茂基双核椅状钴氮配合物,晶体结构采用SHELXS-97程序由直接法解出,结构精修采用SHELXL-97程序,对氢原子和非氢原子分别采用各向同性和各向异性温度因子进行全矩阵最小二乘法修正;最终偏离因子R1=0.0469,wR2=0.1102;w=1/[S2(F02)+(0.1383P)2+1.5640PP],其中P=(F02+2Fc2)/3;(Δ/σ)max=0.000;S=1.064。
本发明的茂基双核椅状钴氮配合物由两个独立的单元结构及5个水分子组成,每个单元结构中由2个Co(II)离子、2个BCNA分子及2个1,10-邻菲啰啉分子组成。Co(II)离子的配位原子主要来自双环[2.2.1]-2-庚烯-5,6-二甲酸离子的羧酸氧原子和1,10-邻菲啰啉分子中的氮原子。钴离子处于五配位的四方锥配位环境中,每个独立单元由2个钴离子2个氧原子构成一个平面四边形结构,两端分别与BCNA分子构成一个椅状结构的化合物,且配合物分子中均存在大量的氢键作用,主要体现在双环[2.2.1]-2-庚烯-5,6-二甲酸中未配位的羧基氧原子与配位水分子中和未配位水分子中的氧原子之间存在丰富的氢键作用,游离水分子之间存在丰富的氢键作用。综上所述,这种钴氮配合物由于具有椅状的茂基结构,分子结构稳定性特别好,在催化酯化反应过程中不易发生结构的异裂,为其稳定、高效的催化能力提供了结构保障,同时相对现有的金属镍配合物催化剂,本发明的茂基双核椅状钴氮配合物在催化效果上具有明显的优势,当催化反应体系达到相同酸值时,采用茂基双核椅状钴氮配合物能明显提高聚合物的聚合程度。
本发明还提供了一种茂基双核椅状钴氮配合物的制备方法,该方法是将双环[2.2.1]-2-庚烯-5,6-二甲酸、钴盐和1,10-邻菲啰啉按摩尔比(1~3):(2~4):(2~5)溶解在混合溶剂中,并调节pH值为6~8,在50~90℃下反应,反应完成后,冷却到室温,过滤,所得滤液在通过自挥发结晶,即得。
本发明的茂基双核椅状钴氮配合物的制备方法还包括以下优选方案:
优选的方案,钴盐包括硫酸钴、碳酸钴、氯化钴、氢氧化钴、硝酸钴、碱式碳酸钴、乙酸钴中至少一种。
优选的方案,混合溶剂主要为不同溶解极性、不同挥发性的溶剂的混合;混合溶剂包括甲醇、乙醇、三氯甲烷和乙腈中的至少一种,与N,N’-二甲基甲酰胺、N,N’-二甲基乙酰胺和水中的至少一种按体积比(1~5):1(更优选的体积比例为(1~4):1)的组合。
较优选的方案,混合溶剂为乙醇和水组合、甲醇和水组合、甲醇和DMF组合、甲醇和N,N’-二甲基乙酰胺组合、乙醇和N,N’-二甲基甲酰胺组合或乙醇和N,N’-二甲基乙酰胺组合。
优选的方案,自挥发结晶是将滤液盛在敞口容器中,用带微孔的保鲜膜将容器口封闭,使溶剂在室温条件下缓慢挥发,直到析出大量晶体。
优选的方案,反应的时间为8~13h。
优选的方案,采用氢氧化钠、氢氧化钾、氨水,三乙胺、二乙胺、三乙醇胺中的至少一种调节pH值。
本发明还提供了一种茂基双核椅状钴氮配合物的应用,将茂基双核椅状钴氮配合物作为催化剂应用于催化酯化反应。
优选的方案,将茂基双核椅状钴氮配合物应用于催化二元酸和二元醇合成聚酯。
优选的制备方法是将双环[2.2.1]-2-庚烯-5,6-二甲酸、钴盐和1,10-邻菲啰啉按摩尔比1~3:2~4:2~5溶解在混合溶剂中,用碱液调节pH值为6~8,在50~90℃下反应8~13h,反应完成后,冷却到室温,过滤,将滤液盛在敞口容器中,用带微孔的保鲜膜将容器口封闭,使溶剂在室温条件下缓慢挥发,直到析出大量晶体。
所述的保鲜膜为常规的市售保鲜膜。
相对现有技术,本发明的技术方案带来的有益效果:
1)本发明的茂基双核椅状钴氮配合物以双环[2.2.1]-2-庚烯-5,6-二甲酸为主配体,以1,10-邻菲啰啉为辅配体,具有特殊的茂基双核椅状结构,其分子结构特别稳定。
2)本发明的茂基双核椅状钴氮配合物对酯化反应表现出较好的稳定性和催化反应活性,特别适用于线性聚酯的合成。
3)本发明的茂基双核椅状钴氮配合物采用一步法合成,装置简单、操作简便,且反应条件温和,在70℃左右即可,且产率较高;此外,采用自挥发结晶,得到的产物颗粒细小均匀,分离提纯容易。
附图说明
【图1】为本发明制备的{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O的晶体结构示意图。
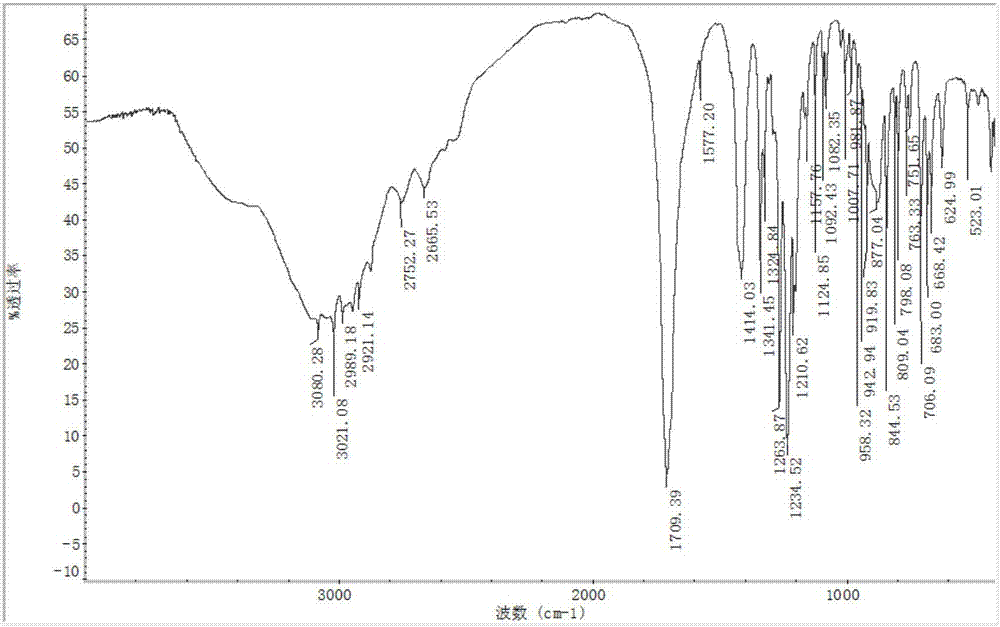
【图2】为本发明采用的双环[2.2.1]-2-庚烯-5,6-二甲酸的红外光谱图。
【图3】为本发明制备的{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O的红外光谱图。
具体实施方式
以下实施例旨在进一步说明本发明的内容,而不是限制本发明权利要求的保护范围。
双环[2.2.1]-2-庚烯-5,6-二甲酸的合成借鉴现有的合成方法:
在125mL三口烧瓶中加入8g马来酸酐和26mL乙酸乙酯,用热水浴加热使固体物全部溶解,然后加入26mL石油醚60~90℃。冷却到室温后,再用冰水浴冷却,再加入6ml新蒸馏的环戊二烯,将反应置于冰水浴中并不断搅拌,直到白色固体析出,得到白色针状结晶,抽滤用15mL乙酸乙酯与石油醚的混合液淋洗,抽滤称重,得到12g双环[2.2.1]-2-庚烯-5,6-二甲酸酐,产率为84%左右,熔点为163.5~165.5℃。将12g产品加入35mL浓度为20%的氢氧化钠溶液中,加热回流1.5小时后溶液由无色变成黄色,2小时后停止加热并冷却至室温,转移到100mL烧杯中趁热过滤,冷却,滤液用浓盐酸酸化到pH值为2~3,抽滤,干燥得到白色针状晶体,10g,产率84%。熔点为167~168℃
IR主要吸收峰(KBr,cm-1)为:3423(s),1717(vs),1643(s),1417(m),1341(s),1327(s),1265(m),1235(vs),1202(w),1158(w),1094(w),1082(w),903(w),844(s),798(w),764(w),751(s),705(w),683(w),665(w),625(w),483(w)。双环[2.2.1]-2-庚烯-5,6-二甲酸元素分析结果按C9H10O4(%),计算值:C,59.34;H,5.53;实验值:C,59.16;H,5.51。1H NMR(400MHz,CDCl3,TMS,δppm):9.6,2.0,2.26,5.6,1.7(-CH),1.8(-CH2),1.06(-CH3)。13CNMR(400MHz,CDCl3,TMS,δppm):14.9,31.8,43.8,43.9,135.8,173.0。
实施例1
茂基双核椅状钴氮配合物{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O的合成,其反应步骤如下:
将0.32mmol(58.24mg)双环[2.2.1]-2-庚烯-5,6-二甲酸、0.34mmol(95.54mg)五水硫酸钴和0.48mmol(95.15mg)1,10-邻菲啰啉加入15mL乙醇和水的混合溶剂中(体积比4:1),用0.2mol/L氢氧化钠溶液调节pH值为6~8,在70℃下反应12h,反应完成后,得到红色的溶液,过滤,滤液上覆盖一层保鲜薄膜,置于室温下自然挥发,20天后析出适合于X射线单晶结构分析的红色晶体,产率48.75%。M.P.185~187℃;IR主要吸收峰为:3416(m),1749(s),1573(vs),1516(m),1425(vs),1406(m),1303(m),1223(w),1182(w),853(vs),784(w),730(vs),639(s)。元素分析C21H21CoN2O6.50:计算值(%):C,55.35;H,4.64;N,6.14;实测值(%):C,53.18;H,4.66;N,6.12;
实施例2
茂基双核椅状钴氮配合物{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O的合成:
将0.54mmol(98.28mg)双环[2.2.1]-2-庚烯-5,6-二甲酸、0.48mmol(84.97mg)四水合醋酸钴和0.67mmol(132.81mg)1,10-邻菲啰啉加入DMF、乙醇和水的混合溶剂中(体积比2:5:1),搅拌溶解,温度控制在65℃,用0.3mol/L的氢氧化钾调节pH值为6.5~7.5,反应10h后,反得到红色的溶液,过滤,滤液上覆盖一层保鲜薄膜,置于室温下自然挥发,25天后析出适合于X射线单晶结构分析的红色晶体,产率46.15%。
实施例3
茂基双核椅状钴氮配合物{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O的合成:
将0.46mmol(83.72mg)双环[2.2.1]-2-庚烯-5,6-二甲酸、0.84mmol(78.08mg)氢氧化钴和0.54mmol(107.04mg)1,10-邻菲啰啉加入甲醇和水的混合溶剂中(体积比5:1),搅拌溶解,温度控制在75℃,用乙二胺调节pH值为6.0~6.5,反应13h后,反得到红色的溶液,过滤,滤液上覆盖一层保鲜薄膜,置于室温下自然挥发,四周后析出适合于X射线单晶结构分析的红色晶体,产率48.4%。
实施例4
对实施1~3制得的{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O晶体进行结构测定
在显微镜下选取尺寸约为0.20×0.18×0.10mm的单晶,置于a Bruker APEX-II CCD单晶衍射仪上进行衍射实验,在173(2)K下用MoKα射线(λ=0.071073nm),以扫描方式在1.54≤θ≤25.02°范围内共收集31323个衍射点,其中7012个独立衍射点[Rint=0.0574],6968个可观察衍射点[I>2σ(I)]用于结构分析和结构修正。全部数据经Lp因子和经验吸收校正。晶体结构采用SHELXS-97程序由直接法解出,结构精修采用SHELXL-97程序,对氢原子和非氢原子分别采用各向同性和各向异性温度因子进行全矩阵最小二乘法修正。最终偏离因子R1=0.0469,wR2=0.1102;w=1/[S2(F02)+(0.1383P)2+1.5640PP],其中P=(F02+2Fc2)/3;(Δ/σ)max=0.000;S=1.064。
配合物分子结构见图1,主要键长和键角列于表1。从晶体结构图1可知,不对称的结构单元中由两个独立的单元结构及5个水分子组成,每个单元结构中包含有2个Co(II)离子、2个BCNA分子及2个1,10-邻菲啰啉分子组成。Co(II)离子的配位原子主要来自双环[2.2.1]-2-庚烯-5,6-二甲酸离子的羧酸氧原子和1,10-邻菲啰啉分子中的氮原子。钴离子处于五配位的四方锥配位环境中,每个独立单元由2个钴离子2个氧原子构成一个平面四边形结构,两端分别与BCNA分子构成一个椅状结构的化合物,中心离子与配体原子之间的键长与键角出现了明显的不同Co-O的键长在范围,Co-N的键长在范围,其平均距离为配合物分子中均存在大量的氢键作用,主要体现在双环[2.2.1]-2-庚烯-5,6-二甲酸中未配位的羧基氧原子与配位水分子中和未配位水分子中的氧原子之间存在丰富的氢键作用,游离水分子之间存在丰富的氢键作用。
表1配合物的主要键长与键角(°)
实施例5
实施例1~3制得的{Co2[C7H8(COO)2]2(C12H8N2)2}·5H2O在聚酯的聚合反应中的催化性能:
称取60g己二酸、25g1,4-丁二醇和15g乙二醇加入到装有温度计、油水分离器和搅拌器的四口烧瓶中,充氮气保护约20min,缓慢加热,物流熔融后开动搅拌器,加入0.1g钴配合物催化剂并逐渐升温,当温度达到145℃左右时开始出水,控制出水速率及顶温在(100±2)℃范围内,同时加大搅拌速率,取样测酸值,当试样酸值低于15mgKOH/g时开始抽真空,于85~90KPa真空度下反应,当酸值小于2.0mgKOH/g时结束反应。冷却至60~80℃出料,得到线性聚酯。其羟值在51~54mgKOH/之间,相对分子质量约为2300。
- 还没有人留言评论。精彩留言会获得点赞!