一种左氧氟沙星异构体化合物的合成方法与流程
本发明涉及医药化学领域,具体涉及到一种(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-7-氧-7h-吡啶并[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸的合成方法。
背景技术:
氟喹诺酮类药物以其广谱、高效、低毒的抗菌特性,在临床抗感染治疗上取得了巨大的成功。其中以氧氟沙星、左氧氟沙星、盐酸左氧氟沙星为其中优秀的代表。
日本第一三共制药有限公司开发的左氧氟沙星为市场占有率较广的优秀品种,化学名为:(-)-(s)-3-甲基-9-氟-2,3-二氢-10-(4-甲基-1-哌嗪基)-7-氧-7h-吡啶并[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸),为半水合物,结构如下:
左氧氟沙星的合成工艺已很成熟,报道文献很多,其中最后一步以缩哌反应合成左氧氟沙星,如专利ep368410、us4777253、wo2006009374、wo2006070275、cn1594320,wo2006070275等,合成路线如下:
专利cn102775424指出上述制备工艺中可能会产生位置异构的杂质(1),
杂质(1)化学名:(s)-(-)-10-氟-2,3-二氢-3-甲基-9-(4-甲基-1-哌嗪基)-7-氧代-7h-吡啶并[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸。该专利提供了一种从母液中回收该杂质的制备方法。但是母液中该杂质含量较少,利用此方法无法满足该杂质作为对照品大量制备的要求。同时该专利指出通过化学的方法制备该物质比较困难,成本较高。
为了解决上述的技术瓶颈,本发明提供了一种工艺简单,起始物料易得的合成方法,以满足对该异构体的制备。迄今为止,未见有文献报道该化合物的化学合成方法。
技术实现要素:
本发明的目的在于提供一种化学合成的方法制备左氧氟沙星的异构体(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-7-氧-7h-吡啶并[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸)。本发明合成制备的得到的左氧氟沙星异构体可为左氧氟沙星的质量分析研究提供对照品,从而提升左氧氟沙星的质量标准。
本发明所说的左氧氟沙星异构体简称9-哌嗪左氧氟沙星,结构如下式(i)所示:
本发明所说的左氧氟沙星的异构体的盐指的(i)与酸所成的盐。
本发明的技术方案如下:
一种左氧氟沙星异构体的合成方法,所述的合成方法包括如下步骤:
a)制备化合物m1:以左氧氟环合酯为原料,在浓硫酸、硝酸或者硝酸盐条件下进行硝化反应;
b)制备化合物m2:向化合物m1中加入n-甲基哌嗪进行缩哌反应;
c)制备化合物m3:用还原剂将化合物m2中的硝基还原为氨基;
d)制备化合物m4:将化合物m3中的氨基转换为氢原子;
e)制备左氧氟沙星异构体:将化合物m4在酸性或者碱性条件下进行酯水解;
反应路线如下:
其中,r1为me或et。
所述的步骤a)中,硝酸盐为硝酸钾或硝酸钠。
所述的步骤b)中,还加有溶剂,所述的溶剂选自n,n-二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮、水、乙醇或乙腈。优选溶剂选自n,n-二甲基甲酰胺,n-甲基吡咯烷酮或水。
所述的步骤c)中,还原剂为fe/nh4cl、zn/nh4cl、fe/乙酸、zn/乙酸或保险粉。优选还原剂为zn/乙酸。
所述的步骤d)中,使用亚硝酸盐、硫酸和还原剂将氨基转换为氢原子;亚硝酸盐为亚硝酸钾或亚硝酸钠;还原剂为乙醇或亚磷酸,优选还原剂为乙醇。
所述的步骤e)中,酸为硫酸或盐酸;碱为氢氧化钠、氢氧化钾、甲醇钠或乙醇钠。
可将所述的步骤e)制备的左氧氟沙星异构体继续制备成盐,所述的盐为乙酸盐、盐酸盐或三氟乙酸盐。
所述的步骤a)中,左氧氟环合酯与硝酸盐的摩尔比为1:1.5;
所述的步骤b)中,m1与n-甲基哌嗪的摩尔比为1:3。
其中,参与反应的各化合物的化学命名为:
左氧氟环合脂:(-)-(s)-3-甲基-9,10-二氟-2,3-二氢-8-硝基-7-氧-7h-吡啶并[1,2,3-de]-1,4-苯并恶嗪-6-羧酸乙(甲)酯
化合物m1:(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-8-硝基-7-氧-7h-吡啶并[1,2,3-de]-1,4-苯并恶嗪-6-羧酸乙(甲)酯
化合物m2:(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-8-硝基-7-氧-7h-吡啶并[1,2,3-de]-1,4-苯并恶嗪-6-羧酸乙(甲)酯
化合物m3:(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-8-氨基-7-氧-7h-吡啶并[1,2,3-de]-1,4-苯并恶嗪-6-羧酸乙(甲)酯
化合物m4:(-)-(s)-3-甲基-10-氟-2,3-二氢-9-(4-甲基-1-哌嗪基)-7-氧-7h-吡啶并[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸乙(甲)酯。
本发明具有如下技术效果:本发明提供了一种工艺简单,起始物料易得的合成方法,可以满足对该异构体的制备。本发明合成制备的得到的左氧氟沙星异构体可为左氧氟沙星的质量分析研究提供对照品,从而提升左氧氟沙星的质量标准。
附图说明
图1是左氧氟沙星异构体式(i)化合物的hplc图。
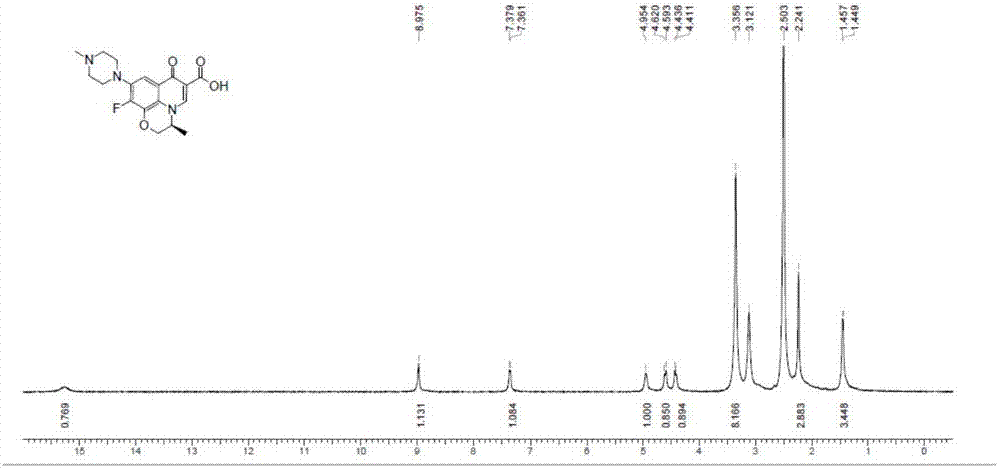
图2是左氧氟沙星异构体式(i)化合物的1hnmr图。
具体实施方式
下面结合实施例对本发明做进一步阐述,本领域技术人员能够理解,这些实施例仅用于说明本发明,但是这些实例不对本发明构成任何限制。
实施例1化合物m1制备
向250ml三口瓶中加入(-)-(s)-9,10-二氟-2,3-二氢-3-甲基-7-氧-7h-吡啶并[1,2,3-de〕-1,4-苯并恶嗪-6-羧酸乙酯4.5g(14.5mmol)、浓硫酸10ml,控制反应温度不超过0℃,缓慢加入硝酸钾2.21g(21.8mmol)。保持温度在0~25℃反应2小时,反应完毕后,向反应液中加入100ml水,固体析出并过滤,干燥后得产物4.8g,淡黄色固体,收率93%。
实施例2化合物m1的制备
向250ml三口瓶中加入(-)-(s)-9,10-二氟-2,3-二氢-3-甲基-7-氧-7h-吡啶并[1,2,3-de〕-1,4-苯并恶嗪-6-羧酸甲酯4.0g(14.5mmol)、浓硫酸10ml,控制反应温度不超过0℃下,缓慢加入硝酸钠1.68g(21.8mmol)。保持温度在0~25℃反应1小时,lcms监控反应完毕后,向反应液中加入100ml水,固体析出并过滤,干燥后得产物4.5g,淡黄色固体,收率87%。
实施例3化合物m2的制备
向250ml三口瓶中加入3.4g化合物m1(9.6mmol),dmf20ml,n-甲基哌嗪2.88g(28.80mmol)。加热至30℃并在此温度下保持12小时,反应完成后向反应液中倒入水100ml,过滤并用水洗涤滤饼,真空干燥后得到产物3.6g,淡黄色固体,收率86%.
实施例4化合物m3的制备
向250ml三口瓶中加入1.6g化合物m2(3.68ml),无水乙醇50ml,水50ml。然后加入铁粉0.82g(14.73mmol),氯化铵0.78g(14.73mmol).加热升温至85℃反应两小时,tlc(二氯甲烷/甲醇=10:1)显示反应完成,过滤催化剂,滤饼用二氯甲烷洗涤,合并滤饼,浓缩至干,快速制备液相色谱分离得到产物,黄色固体0.6g,收率40%。
实施例5化合物m3的制备
向250ml三口瓶中加入1.6g化合物m2(3.68mmol),无水乙酸50ml,乙醇50ml。然后加入锌粉0.93g(14.73mmol),.加热升温至60℃反应两小时,tlc(二氯甲烷/甲醇=10:1)显示反应完成,过滤催化剂,滤饼用二氯甲烷洗涤,合并滤饼,浓缩至干,快速制备液相色谱分离得到产物,黄色固体1.3g,收率86%。
实施例6化合物m4的制备
在0℃下,向100ml三口瓶中加入1.2g化合物m3(2.96mmol),硫酸12ml,亚硝酸钾0.5g。保持0℃下反应1小时,然后加入乙醇15.8g(684mmol),升温至70℃搅拌反应2小时,反应完毕后,减压蒸除乙醇,粗品作为黄色油状物2.5g,不纯化直接投入下步使用。
实施例7左氧氟沙星异构体的制备
在100ml三口瓶中加入2.0g化合物m4,硫酸5ml,水50ml,加热至80℃搅拌12小时,反应完毕后用1mol/l的氢氧化钠溶液调节ph至6.5~7.5,二氯甲烷萃取3次,合并有机相,柱层析(二氯甲烷/甲醇)得到产品1.2g,类白色固体,收率55%。检测结果如图1、图2。
图1是左氧氟沙星异构体式(i)化合物的hplc图。
图2是左氧氟沙星异构体式(i)化合物的1hnmr图(d6-dmso,400mhz)。
从图1,图2可以看出,制备的左氧氟沙星异构体化合物纯度较高(>99%),可以满足其作为对照品及其药理毒理研究的纯度要求
实施例8左氧氟沙星异构体乙酸盐的制备
在100ml三口瓶中加入左氧氟沙星异构体1.0g,水10ml,加入乙酸1.0g,搅拌0.5小时后加入乙醇20ml,过滤,得左氧氟沙星异构体的乙酸盐0.9g。
实施例9左氧氟沙星异构体盐酸盐的制备
在100ml三口瓶中加入左氧氟沙星异构体1.0g,水10ml,加入盐酸3ml,搅拌0.5小时后加入乙醇20ml,过滤,得左氧氟沙星异构体的盐酸盐0.8g。
- 还没有人留言评论。精彩留言会获得点赞!