一种分子基磁性化合物、晶体及其制备方法和用途与流程
本发明属于分子基磁性材料技术领域,具体涉及一种具有[ni(dmit)2]阴离子和苄基喹啉阳离子的分子基磁性化合物及其晶体,同时还涉及该化合物和晶体的制备方法和用途。
背景技术:
自1967年wickman发现第一个分子基磁体以来,尤其是miller和kahn等人报道分子铁磁体以来,分子基磁性材料已经成为目前物理、化学和材料等多个领域的研究热点和前沿课题之一。与传统的单原子或离子为基体的合金或氧化物磁体不同的是,分子基磁性材料以分子为构件,其磁性来源于分子间的磁耦合相互作用,在临界温度下呈现出三维磁有序化的排列方式,故而分子基磁性材料具有传统磁性材料无法比拟的优点,例如体积小、密度轻、结构多样化、易于复合加工成型等。
分子基磁性材料由分子基磁性化合物构成,分子基磁性化合物具有分子合成方式的无限性和分子结合方式的多样性等特点,自旋转换过渡金属配合物是其中最为重要的一类分子基磁性化合物。在上述配合物中,中心金属离子在正八面体场中具有两种电子排列方式(高自旋态和低自旋态),在合适的条件下高低自旋态之间可以互相转变。利用这种特殊的自旋双稳性,可以在分子水平上实现光开关、热开关和信息存储材料的应用。在现有的自旋转换过渡金属配合物中,双-1,2-二硫烯化合物由于其优异的导电和磁学性质受到了广泛关注。这类化合物具有特殊的几何构型以及分子间的相互作用,其平面型的阴离子包含金属离子和二硫杂环戊烯单元,绝大部分的负电荷都集中在该平面型的骨架上。它能与苄基氮杂环阳离子形成阴阳离子完全分列的柱状堆积结构,通过系统地改变阳离子的分子构象,调节阳离子的分子拓扑和体积大小,进一步调控自由基分子阴离子的堆积结构,从而获得具有分子双稳性的一维分子基磁性材料。
例如,陈存友等人采用dmit(coph)2和苄基吡啶盐[ncbzpy]cl为原料,合成了一种分子基磁性化合物[ncbzpy][ni(dmit)2]。然而,该磁性化合物的高低自旋态之间的转变温度(即居里点温度)非常低,距离可实际应用的温度相差甚远。不仅如此,现有的其它dmit系分子基磁性化合物的居里点温度也都较低,并且高低自旋态之间转变的热滞后环的温度范围均较窄,使其难以满足分子电子器件的实用性要求。因此,设计合成具有更高居里点温度和更宽热滞后环温度范围的分子基磁性材料,已成为当前分子磁学和材料科学领域面临的极大挑战。
技术实现要素:
本发明所要解决的技术问题在于克服现有的分子基磁性化合物的居里点温度较低且高低自旋态之间转变的热滞后环的温度范围较窄的缺陷,进而提供一种具有更高居里点温度和更宽热滞后环温度范围的分子基磁性化合物、晶体及其制备方法和用途。
为此,本发明实现上述目的的技术方案为:
一种分子基磁性化合物,具有如式(i)所示的结构:
其中,r1和r2各自独立地为h或吸电子基团。
优选地,所述吸电子基团为no2、cn、cf3、卤素、cooh、so3h或coor3,其中r3为c1-c10烷基。
更优选地,所述r1和r2均为h;该化合物的晶体在x射线衍射图谱中包含以下2θ角的特征峰:7.0±0.2°、10.5±0.2°、12.0±0.2°、12.5±0.2°、18.0±0.2°、18.5±0.2°、23.3±0.2°、26.0±0.2°和31.5±0.2°。
进一步地,一种制备上述分子基磁性化合物的方法,包括下列步骤:
将溶解有[bu4n][ni(dmit)2]的有机溶剂缓慢滴加至溶解有苄基喹啉盐的有机溶剂中以发生反应,待反应完成后过滤,收集滤饼,干燥,即得所述分子基磁性化合物;
其中,所述[bu4n][ni(dmit)2]的结构如式(ii)所示:
所述苄基喹啉盐的结构如式(iii)所示:
式(iii)中,x代表卤素。
优选地,所述[bu4n][ni(dmit)2]与所述苄基喹啉盐按摩尔比1:(1~1.2)混合,并于室温下搅拌反应6~24小时。
优选地,所述有机溶剂为乙腈。
更优选地,还包括采用丙酮对所述滤饼进行重结晶的步骤。
进一步地,上述分子基磁性化合物或上述制备方法制得的分子基磁性化合物在制备分子基磁性材料中的用途。
一种分子基磁性材料,由上述分子基磁性化合物或上述制备方法制得的分子基磁性化合物制备而成。
上述分子基磁性材料作为光开关材料、热开关材料或信息存储材料的应用。
与现有技术相比,本发明的上述技术方案具有以下有益效果:
1、本发明提供的分子基磁性化合物,具有[ni(dmit)2]阴离子和苄基吡啶阳离子,该化合物的高低自旋态在升温过程中的转变温度较高,其居里点温度可达250k,与现有的其它分子基磁性化合物相比更接近于室温,故而使得由该化合物制备得到的分子基磁性材料可在接近室温的条件下使用。
并且本发明所述的分子基磁性化合物的高低自旋态之间转变的热滞后环的温度范围更宽,高达200k以上,使得由其制备得到的分子基磁性材料可在较宽温度范围下应用。
2、本发明提供的分子基磁性化合物的制备方法,通过将溶解有[bu4n][ni(dmit)2]的有机溶剂缓慢滴加至溶解有苄基喹啉盐的有机溶剂中以发生反应,由此可显著提高目标产品的收率。
附图说明
为了更清楚地说明本发明具体实施方式中的技术方案,下面将对具体实施方式描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
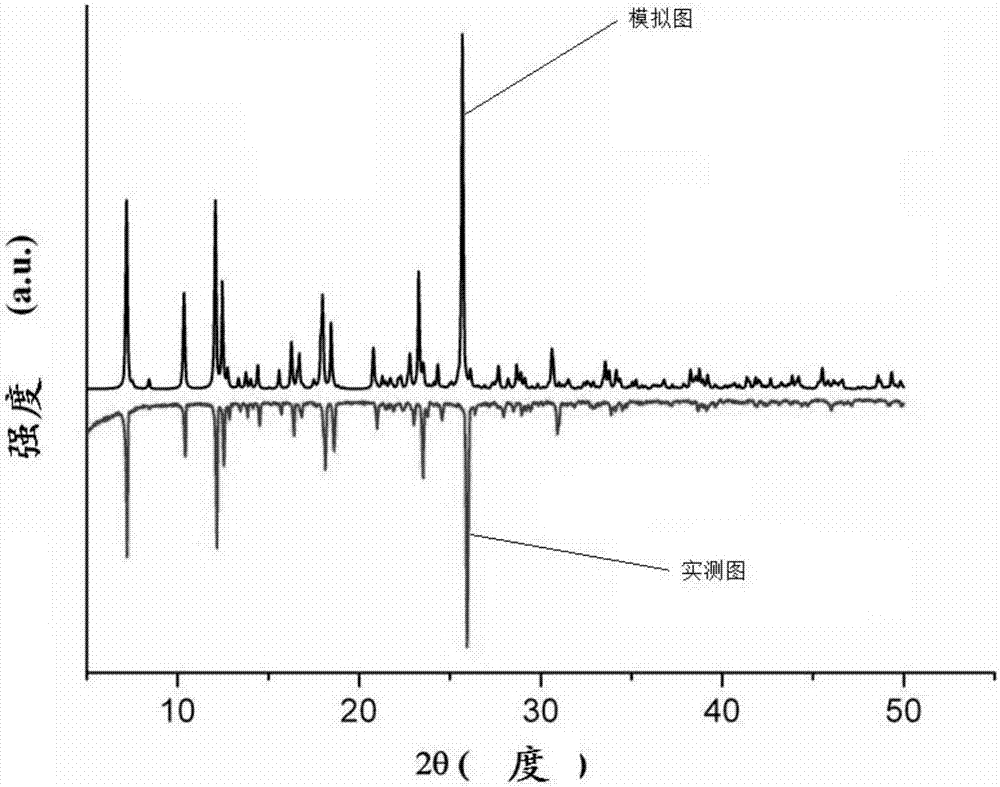
图1为实施例1制得的化合物的x-射线粉末衍射图;
图2为实施例1制得的化合物沿着b轴方向的堆积结构图;
图3为实施例1制得的化合物的变温摩尔磁化率曲线图;
图4为实施例1制得的化合物的单晶结构图。
具体实施方式
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
下述实施例中所用试剂均为分析纯。
实施例1
本实施例提供了一种具有如式(i-a)所示结构的分子基磁性化合物的制备方法,包括:
(1)将[bu4n]2[zn(dmit)2]与phcocl以化学计量比为1:3混合并搅拌均匀,抽滤,滤饼用体积比为1:1的乙醇和丙酮的混合溶剂洗涤,晾干,将所得固体在氮气保护下溶解于甲醇中并使之质量分数为2%,而后加入质量分数为1%的ch3ona的甲醇溶液,搅拌均匀后再加入质量分数为0.5%的六水合氯化镍的甲醇溶液,其中上述溶解于甲醇中的固体与ch3ona、六水合氯化镍的摩尔比均为0.5,再次搅拌均匀后加入四丁基溴化铵,控制六水合氯化镍与四丁基溴化铵的化学计量比为0.5:2,抽滤,滤饼用甲醇洗涤,晾干,将所得固体溶解于丙酮中,加入质量比为15:2的ki与i2的混合物,搅拌以均匀混合,其中ki与i2的混合物与溶解有上述固体的丙酮溶液之间的摩尔比例关系是1:1;而后在10℃下自然挥发溶剂,得到[bu4n][ni(dmit)2];上述反应方程式如下:
(2)将693mg的[bu4n][ni(dmit)2]溶解于15ml乙腈中,再将此乙腈溶液缓慢滴加到溶解有350mg[bzql]br的15ml乙腈溶液中,室温下搅拌12小时,过滤分离,得到黑色沉淀,沉淀物用乙腈洗涤后置于空气中干燥,得到式(i-a)所示结构的分子基磁性化合物[bzql][ni(dmit)2],产率为82%;
(3)使用丙酮对步骤(2)所得[bzql][ni(dmit)2]进行重结晶,即得到该化合物的晶体。
采用elementarvarioeliii元素分析仪对本实施例所得化合物i-a进行元素分析。该化合物的c、h、n三种元素含量的实际测量值与计算值列于表1。
表1本实施例制得的化合物i-a的元素分析数据
由表1可知,本实施例制得的化合物i-a在实验误差范围内的实测值与其计算值一致。
采用溴化钾压片法在170sx傅里叶转换红外光谱仪上对本实施例所得化合物i-a进行红外光谱测定(4000-400cm-1),红外光谱带的主要特征峰如下所示:
3051.68w(苯环和喹啉环上c-h(νc-h)伸缩振动);
2929.79w(-ch2-亚甲基的c-h(νc-h)伸缩振动);
1620.97w(喹啉环上c-n(νc-n)伸缩振动);
1336.08s(dmit2-配体上c=c(νc=c)的伸缩振动);
1064.35s(dmit2-配体上c=s(νc=s)的伸缩振动);
756.89m(dmit2-配体的c-s-c(νc-s-c)伸缩振动);
506.79m(dmit2-配体c-s(νc-s)伸缩振动)。
采用brukerd8-advanced粉末x-射线衍射仪对本实施例所得的化合物i-a的晶体进行测定。实验条件为:x-射线管电压40kv,电流30ma,x-射线源为cukα射线,数据收集范围5°﹤2θ﹤50°。化合物i-a的xrd谱图如图1所示,从图1可以看出,化合物i-a的x射线粉末衍射实测图与模拟图一致,由此进一步表明,化合物i-a的晶体产物具有很高相纯度。
化合物i-a的晶体的x-射线单晶数据收集和结构解析如下:
室温时,在brukersmartapexccd上,用石墨单色器单色化的mo靶kα射线
表2本实施例制得的化合物i-a的晶体数据和结构精修参数
如图2所示,在此晶体中,沿着b轴方向,通过侧边的s…s堆积,[ni(dmit)2]-阴离子形成均匀链(较短s…s距离:
本实施例制得的化合物i-a的变温摩尔磁化率曲线如图3所示。在300k时,其χmt值为0.319emu·k·mol-1,与理论值s=1/2自旋体系唯自旋值0.375emu·k·mol-1相近。在250-300k之间,χm值几乎保持不变;当温度低于250k后,χm值开始降低并在140k时达到最小值。此后,随着温度继续降低,χm迅速增大并在40k时达到最大值;在2-30k范围内的低温区,磁化率随着温度的降低急剧增大,出现了居里尾。在紧接着升温过程中,2-30k范围内,降温和升温磁化率曲线重合。但是,在30-256k温区出现了一个形状罕见的热滞回线。显然,在降温过程中,该化合物于140k附近开始由顺磁向抗磁发生转变;在升温过程中,在250k左右发生了高自旋态向低自旋态的转变。
实施例2-10
按照与实施例1相同的制备方法,但将其中的反应物[bzql]br更换为其它合适的苄基喹啉盐,从而制备实施例2-10的化合物,并且测定其变温摩尔磁化率曲线,记录这些化合物的热滞后环的温度范围、降温过程中磁相变温度以及升温过程中的磁相变温度。分别参见下表3。
表3实施例2-10制得的化合物的变温摩尔磁化率数据
综上所述,本发明所述的分子基磁性化合物具有自旋双稳性。高低自旋态在升温过程中的转变温度较高,接近室温条件;同时高低自旋态之间转变的热滞后环的温度范围高达200k以上,因此可以在较宽工作温度下应用。本发明的化合物尤其可以用于制备在分子水平上实现热开关、光开关、信息存储等功能的磁性材料。
对比例1
本对比例提供的一种具有如式(i-a)所示结构的分子基磁性化合物的制备方法,包括:
按照实施例1的方法制备[bu4n][ni(dmit)2],而后向30ml乙腈中加入693mg的[bu4n][ni(dmit)2]和350mg的[bzql]br,室温下搅拌12小时,过滤分离,得到黑色沉淀,沉淀物用乙腈洗涤后置于空气中干燥,得到式(i-a)所示结构的分子基磁性化合物[bzql][ni(dmit)2],产率为75%。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!
- 过渡金属硫属化合物中空多孔纳米片的制备方法及其应用与流程
- 过渡金属二硫属化合物光热响应型多重形状记忆膜材料及其制备方法与流程
- 从亚铁磁稀土过渡金属(RE‑TM)合金形成的自旋转移切换磁性元件的制作方法与工艺
- 一种负载过渡金属硫族化合物的氮功能化碳材料及其制备和应用的制作方法与工艺
- 过渡金属化合物、含有所述化合物的催化剂组合物及利用所述组合物的聚合物的制备方法与流程
- 新颖的第4族过渡金属化合物及其用途的制造方法与工艺
- 新颖的第4族过渡金属化合物及其用途的制造方法与工艺
- 水溶性盐辅助转移CVD二维过渡金属硫族化合物的方法与流程
- 芳族化合物的无过渡金属的甲硅烷基化的制造方法与工艺
- 过渡金属化合物以及包含该过渡金属化合物的催化剂组合物的制造方法与工艺